Resumen
Los pulmones constituyen un entorno inmunológicamente único; están expuestos diariamente a innumerables patógenos y partículas. La eliminación adecuada de patógenos y la respuesta a los contaminantes son esenciales para prevenir infecciones graves, a la vez que se evita el daño tisular y se mantiene un intercambio gaseoso eficiente. En términos generales, el sistema inmunitario innato comprende el conjunto de respuestas inmunitarias intrínsecas e inmediatas ante patógenos o lesiones tisulares. En esta revisión, examinaremos las respuestas inmunitarias innatas del pulmón, con especial atención a su papel en la neumonía. Analizaremos las barreras anatómicas y las proteínas antimicrobianas del pulmón, el reconocimiento de patógenos y lesiones, y la función de los leucocitos (macrófagos, neutrófilos y linfocitos innatos) y las células estromales pulmonares en la inmunidad innata. A lo largo de la revisión, nos centraremos en los nuevos hallazgos sobre la inmunidad innata, así como en las características exclusivas del pulmón.
Introducción
La neumonía es un problema de salud pública importante, que causa una morbilidad y mortalidad significativas anualmente, incluso antes de la pandemia de COVID-19. Clínicamente, la neumonía se define por un conjunto de síntomas (tos, fiebre, dificultad para respirar), además de un nuevo infiltrado en la radiografía de tórax. Dentro de esta definición amplia, la gravedad de la enfermedad puede variar de leve a grave, pudiendo complicarse con sepsis, bacteriemia, shock, síndrome de dificultad respiratoria aguda (SDRA) y disfunción multiorgánica. La neumonía puede ser causada por múltiples patógenos, incluyendo bacterias grampositivas y gramnegativas, virus y hongos. A pesar de la amplia gama de agentes causales, existen muchas vías comunes en las respuestas inmunitarias a la neumonía, lo que sugiere que la disfunción inmunitaria del huésped es un determinante subyacente de la neumonía grave.
El sistema inmunitario innato es ancestral y muchos de sus aspectos se conservan en vertebrados, invertebrados y plantas. En cierto sentido, puede definirse como aquellos aspectos de la defensa del huésped codificados en la línea germinal que no requieren la reorganización genética central de la inmunidad adaptativa. Funcionalmente, los miembros de la respuesta inmune innata reconocen patrones moleculares conservados, activan mecanismos de defensa inmediatos, alertan y reclutan a otros miembros de la respuesta inmune al sitio de infección y coordinan las respuestas con la respuesta inmune adaptativa. Este sistema incluye barreras físicas, mecanismos de reconocimiento de patrones, la eliminación de células por parte de células inmunes y proteínas antimicrobianas, y la coordinación mediante la señalización de citocinas (Fig. 1). En esta revisión, describiremos las características de la respuesta inmune innata, en particular su relación con el pulmón y su papel en la neumonía.
Defensa del huésped en el contacto inicial
Barreras anatómicas
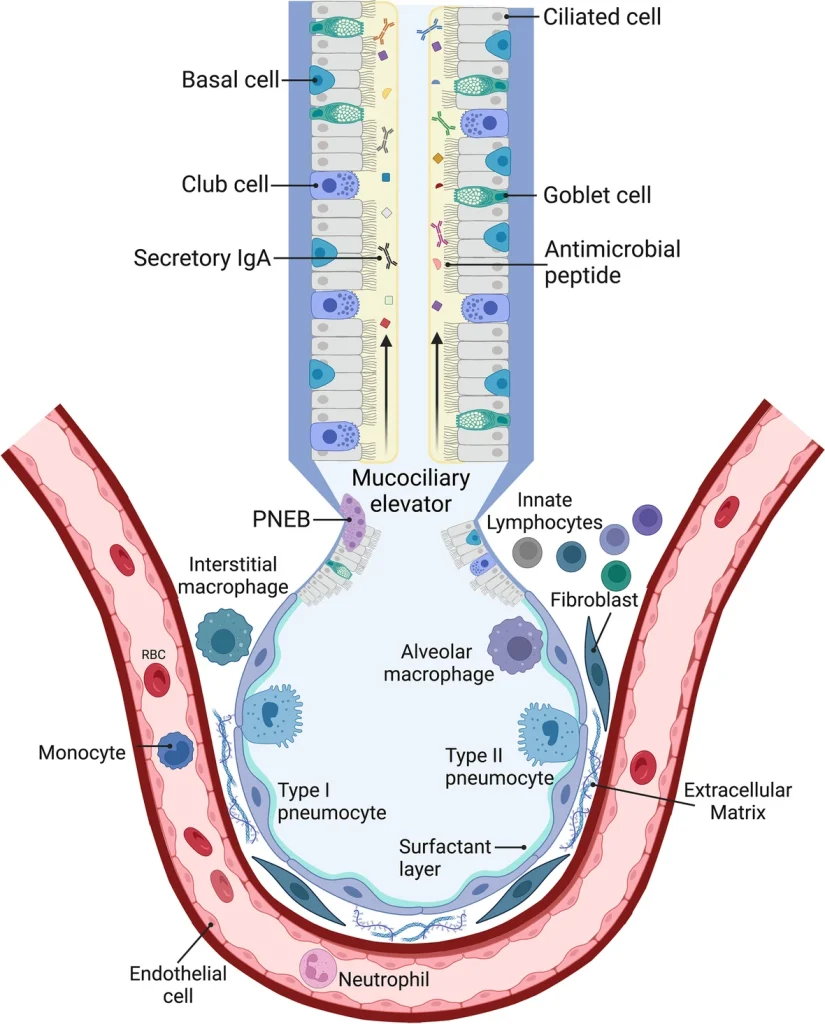
El pulmón humano promedio inhala aproximadamente 11 000 L de aire al día, el cual contiene una mezcla de gases, partículas finas (como polen y detritos) y microorganismos. Por ello, el pulmón posee diversas barreras anatómicas que constituyen la primera línea de defensa contra los patógenos pulmonares. Las células epiteliales de las vías respiratorias conductoras forman una barrera física y química continua, compuesta por epitelio pseudoestratificado en las vías respiratorias proximales y epitelio cúbico simple en los bronquiolos más distales. Si bien su función es heterogénea, todas las células epiteliales del pulmón comparten la capacidad de regular la permeabilidad paracelular mediante uniones estrechas y adherentes. Estas proteínas, ampliamente descritas en otras publicaciones, proporcionan una barrera física entre el espacio aéreo y el intersticio a través de sus conexiones con las proteínas del citoesqueleto de las células vecinas. Estas interacciones paracelulares previenen la diseminación de microorganismos y la fuga vascular, procesos que pueden verse alterados durante una lesión inflamatoria por factores del huésped como el TNF-α (factor de necrosis tumoral alfa) y toxinas derivadas de patógenos, como la neumolisina y el lipopolisacárido.
La acción coordinada de las células ciliadas y caliciformes de las vías respiratorias superiores también proporciona una función esencial para la defensa anatómica: la depuración mucociliar. Las vías respiratorias superiores están revestidas por un líquido superficial complejo, compuesto por una capa superior gelatinosa de moco que atrapa físicamente los microbios y partículas del ambiente, y una capa inferior más fluida que facilita el movimiento de los cilios. Si bien contiene más de 200 proteínas, la capa de moco está compuesta principalmente por mucinas, en particular MUC5AC y MUC5B (mucina 5AC y 5B), secretadas por las células caliciformes y las glándulas submucosas. Recientemente revisadas en detalle, estas glicoproteínas poliméricas contribuyen a la composición física del líquido de revestimiento de las vías respiratorias, lo que promueve la depuración mucociliar de más del 90 % de las partículas inhaladas. Además, se sabe que las mucinas tienen una función antimicrobiana directa. La MUC5B es especialmente crucial para la regulación homeostática de la depuración mucociliar, cuya deficiencia conduce a una infección crónica y a una reducción de la función de los macrófagos y de la producción de interleucina-23 (IL-23). Por otro lado, la MUC5AC es importante en la defensa del huésped contra el virus de la influenza, posiblemente a través de sus interacciones con el ácido siálico α2,3-ligado. Curiosamente, la MUC1 (mucina 1), una mucina transmembrana expresada tanto por células epiteliales como inmunitarias, tiene una función inmunomoduladora al atenuar la señalización inflamatoria durante la infección respiratoria mediante la interacción directa con los receptores tipo Toll (TLR). Recientemente se ha demostrado que el aumento de la secreción de mucina tras una infección por influenza o Streptococcus pneumoniae depende parcialmente de la señalización del interferón de tipo I, cuya producción también se ha asociado con un aumento de la diseminación y transmisión microbiana.
Proteínas antimicrobianas
Además de las mucinas, el líquido que recubre las vías respiratorias contiene proteínas adicionales que contribuyen a la defensa inmediata contra posibles patógenos. Entre estas proteínas se encuentran las inmunoglobulinas generadas a través de las vías inmunitarias adaptativas, que hemos incluido en esta sección para mayor exhaustividad, pero que requieren una infección previa para activar una respuesta de memoria específica. Las células plasmáticas subepiteliales producen inmunoglobulina A secretora (s-IgA), que son dímeros de IgA unidos covalentemente a una glicoproteína denominada componente secretor. Tanto la s-IgA como el componente secretor son transportados por transcitosis al lumen de las vías respiratorias mediante un receptor de inmunoglobulina G (IgG) polimérico ubicado en las células epiteliales pulmonares, lo que proporciona protección de memoria contra la influenza y Streptococcus pneumoniae de forma antígeno-específica. Curiosamente, el componente secretor y la s-IgA, juntos o por separado, también pueden prevenir la infección de forma inespecífica. Esto ocurre mediante la interacción con componentes bacterianos o mediante la inmunomodulación de factores del huésped, como la inhibición de la interleucina-8 (IL-8). La lactoferrina y la lisozima son dos componentes adicionales del líquido de revestimiento epitelial de los pulmones. Se ha demostrado que la lactoferrina, además de secuestrar hierro, desintegra las biopelículas microbianas producidas por Streptococcus pneumoniae, un importante colonizador de la nasofaringe y agente causal de la neumonía adquirida en la comunidad. La lisozima, que degrada enzimáticamente la estructura del peptidoglicano de las bacterias, contribuye directamente a la defensa inmunitaria pulmonar contra patógenos tanto grampositivos como gramnegativos. Además de los mencionados, otros factores humorales que contribuyen a una defensa rápida contra la infección pulmonar incluyen el complemento y otras proteínas de fase aguda, como la proteína C reactiva, las proteínas amiloides séricas y la pentraxina 3.
Un componente único y fundamental del medio alveolar es el surfactante pulmonar, secretado por las células epiteliales alveolares de tipo II. Fundamentales para reducir la tensión superficial en el pulmón, las proteínas surfactantes A y D (SP-A y SP-D) son colectinas que facilitan la eliminación de patógenos virales y bacterianos mediante una mayor captación, aglutinación o interferencia con su receptor específico, especialmente en el caso de infecciones virales. Recientemente, se ha demostrado que tanto la SP-A como la SP-D son dianas de la elastasa de neutrófilos, un resultado que podría explicar parcialmente la mayor susceptibilidad de los pacientes con deficiencia de alfa-1 antitripsina a las infecciones pulmonares bacterianas. Las proteínas surfactantes también modulan la inflamación a través de sus interacciones con células innatas y adaptativas en el pulmón, cuyos detalles se han revisado en otras publicaciones. Finalmente, se ha identificado la SP-D sérica como un fuerte predictor de la gravedad de la neumonía adquirida en la comunidad en niños, probablemente debido al aumento de la permeabilidad alveolar que se observa comúnmente durante las infecciones pulmonares graves. Otras colectinas, como la lectina de unión a manosa, las ficolinas-1-3 y las colectinas-10-11, revisadas recientemente en detalle, reconocen fracciones de carbohidratos y proporcionan una protección rápida contra una miríada de patógenos respiratorios, principalmente a través de la opsonización mediada por el complemento.
Receptores de reconocimiento de patrones y vías de señalización
Un factor clave de la respuesta inmune innata es la respuesta muy rápida (de minutos a horas) a patógenos o lesiones tisulares. Como se revisó recientemente en otra publicación, esto se facilita mediante la unión de moléculas que contienen patrones moleculares asociados a patógenos (PAMP) o patrones moleculares asociados a daño (DAMP) a sus receptores de reconocimiento de patrones (PRR) correspondientes en la superficie de las células del huésped. En general, los PAMP son motivos moleculares conservados expresados por microorganismos pero no por los tejidos del huésped, mientras que los DAMP son motivos moleculares derivados del huésped que solo se liberan durante una lesión tisular. La unión de PAMP/DAMP a los PRR conduce a la activación de las vías de señalización inmune innata, la producción de citocinas y quimiocinas inflamatorias y el reclutamiento de células efectoras. En esta sección, describiremos los PRR y sus principales vías de señalización posteriores.
Receptores tipo Toll
Los receptores de reconocimiento de patrones más conocidos y mejor caracterizados son los receptores tipo Toll (TLR, Tabla 1), un grupo de proteínas transmembrana ubicadas en la membrana plasmática o endosomal. Sus ectodominios contienen repeticiones ricas en leucina, que también constituyen sus dominios primarios de unión a ligandos. Los ligandos están conservados, varían entre los receptores e incluyen productos microbianos como el lipopolisacárido (LPS, que se une a TLR4), lípidos de membrana microbianos (ácido lipoteicoico, lipoarabinomanano o zimósano, que se unen a los heterodímeros TLR1/2 y TLR2/6), flagelina (que se une a TLR5) y ADN CpG no metilado (que se une a TLR9). La unión y señalización de los TLR se ha revisado en detalle recientemente; por lo tanto, este trabajo se centrará en los avances recientes durante la infección pulmonar. La administración de un cóctel de agonistas de TLR (Pam2-ODN, agonistas de TLR2/6 y 9) durante la infección por paramixovirus Sendai conduce a la atenuación del daño epitelial pulmonar temprano en la infección, reducción de la carga viral y disminución de la mortalidad en ratones. En un estudio observacional, los pacientes ancianos con neumonía grave exhibieron niveles más bajos de expresión de TLR2 y TLR4 en monocitos, correlacionados con concentraciones séricas disminuidas de interleucina-1 (IL-1), interleucina-6 (IL-6) y TNF-α. Además, los pacientes, especialmente los niños, con una deficiencia en MyD88, son mucho más susceptibles a la neumonía, probablemente debido a defectos en la señalización de TLR. Durante la neumonía por S. pneumoniae, la eliminación de TIR8 (receptor Toll/interleucina-1 8), que es un regulador negativo de la señalización de TLR, mejora la eliminación bacteriana y la supervivencia debido a una mayor fagocitosis por leucocitos en el pulmón. Los agonistas de TLR incluso se están considerando como tratamientos contra la neumonía, ya que los agonistas de TLR2/6 y TLR9 han demostrado ser eficaces para reducir la mortalidad cuando se combinan con oseltamivir durante la infección por influenza A en ratones. Además, la combinación de agonistas de TLR5 y antibióticos mejora el pronóstico de la infección, incluso en respuesta a cepas de S. pneumoniae resistentes a los antibióticos. Por el contrario, la señalización de TLR puede ser perjudicial para el pronóstico de la neumonía, especialmente cuando la inmunopatología es el principal factor contribuyente a la enfermedad, como se ha demostrado para TLR3 tras la infección por Klebsiella pneumoniae.
TLR1 |
Lipoproteínas bacterianas |
Membrana plasmática |
Citocinas inflamatorias |
TIRAP/MAL, MyD88 |
TLR2 |
Lipoproteínas bacterianas |
Membrana plasmática |
Citocinas inflamatorias |
TIRAP/MAL, MyD88 |
TLR3 |
ARN de doble cadena (dsRNA) |
Membrana endosomal |
Interferón tipo 1 |
TRAM, TRIF |
TLR4 |
LPS |
Membrana plasmática |
Interferón tipo 1 |
TRAM, TRIF |
TLR5 |
Flagelina |
Membrana plasmática |
Citocinas inflamatorias |
TIRAP/MAL, MyD88 |
TLR6 |
Lipoproteínas bacterianas |
Membrana plasmática |
Citocinas inflamatorias |
TIRAP/MAL, MyD88 |
TLR7 |
ARN de cadena simple (ssRNA) |
Membrana endosomal |
Citocinas inflamatorias |
TIRAP/MAL, MyD88 |
TLR8 |
ARN de cadena simple (ssRNA) |
Membrana endosomal |
Citocinas inflamatorias |
TIRAP/MAL, MyD88 |
TLR9 |
ADN CpG |
Membrana endosomal |
Citocinas inflamatorias |
TIRAP/MAL, MyD88 |
Tabla 1. Receptores tipo Toll (TLR)
Receptores tipo NOD
Los receptores tipo NOD (receptores tipo dominio de oligomerización de unión a nucleótidos, NLR, Tabla 2) son un conjunto diverso de receptores intracelulares que responden a una amplia variedad de PAMP y DAMP. Si bien comparten motivos estructurales conservados, sus mecanismos de activación y función posterior son bastante variados. Algunos NLR, como NLRP3, tienen capacidad de oligomerización para formar inflamasomas que conducen a la activación de vías de muerte celular y a la escisión proteolítica y liberación de interleucina-18 (IL-18) e interleucina-1β (IL-1β). Además, otros productos liberados por células muertas y moribundas, llamados alarminas, incluyen HMGB1, IL-33 e IL-1α. Las alarminas promueven la defensa del huésped y la inflamación a través de diversos mecanismos. HMGB1 interactúa con su receptor RAGE, que se encuentra exclusivamente en altas concentraciones en el pulmón y promueve la señalización proinflamatoria a través de la activación de NF-kB. La IL-33 y la IL-1α son citocinas de la familia IL-1 que actúan a través de sus receptores ST2 e IL-1, respectivamente. La IL-33 protege contra la lesión pulmonar aguda inducida por neumonía, posiblemente mediante un aumento de la eosinofilia y una menor captación de neutrófilos, mientras que el papel de la IL-1α (en comparación con la IL-1β) no se comprende del todo, pero probablemente actúa a través de mecanismos dependientes del hígado.
Subfamilia |
Miembro de la subfamilia |
Ligando |
Función |
Señalización |
NLRA |
CIITA |
IFNγ |
Regulador positivo de la transcripción de MHC II |
Factor de transcripción para MHC |
NLRB |
NAIP |
Flagelina; T3SS |
Antiapoptosis |
Inflamasoma mediado por caspasa-1 |
NLRC |
NOD1 |
Dipeptido iE-DAP |
Citocinas inflamatorias; proteínas antimicrobianas |
RIP2, NFκB, MAPK |
NLRC |
NOD2 |
Dipeptido muramil (MDP) |
Citocinas inflamatorias; proteínas antimicrobianas |
RIP2, NFκB, MAPK |
NLRC |
NLRC3 |
ADN viral y ácidos nucleicos |
Regulación negativa de NFκB; atenúa IFN tipo I |
TRAF6 |
NLRC |
NLRC4 |
Flagelina; T3SS; T4SS |
Formación de inflamasoma |
Caspasa-1 |
NLRC |
NLRC5 |
Nucleósido trifosfato |
Regulación de MHC I |
Transcripción de MHC I |
NLRC |
NLRX1 |
dsRNA, PUA, ESA y DHA |
Regula negativamente IFN tipo I e IL-6; localizado en mitocondria |
NFκB |
NLRP |
NLRP1 |
MDP, toxina letal del ántrax |
Formación de inflamasoma |
Caspasa-1 |
NLRP |
NLRP3 |
PAMPs y DAMPs asociados a infección viral |
Formación de inflamasoma |
Caspasa-1 |
NLRP |
NLRP6 |
Desconocido |
Formación de inflamasoma poco caracterizada |
Regulación negativa de NFκB y MAPK |
NLRP |
NLRP7 |
Lipopéptido |
Formación putativa de inflamasoma |
Desconocido |
NLRP |
NLRP12 |
Desconocido |
Formación de inflamasoma no comprobada |
Regulación negativa de NFκB y MAPK |
Tabla 2. Receptores tipo NOD (NLR)
Los NLR que no generan inflamasomas (NLRC1-3, NLRC5 y NLRX1) desempeñan varias funciones importantes durante la infección, mediando tanto las vías de defensa inmunitaria (proinflamatorias) como las de resolución (antiinflamatorias). Los detalles sobre la activación y señalización de los NLR se revisaron recientemente en otra publicación. La expresión de NLRC4 aumenta tanto en pulmones humanos como de ratones con neumonía. Curiosamente, durante la neumonía experimental con Staphylococcus aureus, los ratones muestran una mayor supervivencia y una menor carga bacteriana con la ablación de NLRC4. Se demostró que los efectos protectores de la deficiencia de NLRC4 se producen a través de la inhibición de la necroptosis y la mejora en el reclutamiento de neutrófilos dependiente de interleucina-17A (IL-17A). De manera similar, la activación del inflamasoma NLRP6 impidió la afluencia de neutrófilos y la actividad antimicrobiana durante la infección por S. aureus, probablemente a través del aumento de las vías de muerte celular inflamatoria (piroptosis y necroptosis) y la reducción del interferón-γ (IFN-γ). Sin embargo, limitar la señalización de NLRP6 no siempre es beneficioso. La deficiencia de NLRP6 durante la infección por K. pneumoniae resulta en una mayor mortalidad y diseminación bacteriana extrapulmonar debido a un defecto en el reclutamiento de neutrófilos y la formación de trampas extracelulares de neutrófilos (NET). Finalmente, los ratones deficientes en NLRP3 o su adaptador asociado ASC (proteína similar a la mancha asociada a la apoptosis que contiene un CARD) demostraron una producción robusta y temprana de citocinas, lo que condujo a una mejor eliminación del serotipo 3 de S. pneumoniae. Si los NLR promueven una respuesta que facilita la eliminación bacteriana parece ser específico del contexto, dependiendo del patógeno, la cepa y/o la carga patógena.
Receptores tipo RIG-I
Los receptores tipo RIG-I (tipo gen inducible por ácido retinoico-I, RLR, Tabla 3) son moléculas sensoras intracelulares que responden al ARN viral y median la producción de interferones tipo I y tipo III. Se conocen tres RLR (RIG-I, proteína 5 asociada a la diferenciación del melanoma (MDA5) y LGP2), cada uno con un dominio C-terminal conservado que reconoce el ARN y un dominio helicasa central. Además, RIG-I y MDA5 contienen dominios de reclutamiento de caspasas (CARD) N-terminales que facilitan su interacción con una proteína adaptadora, MAVS (proteína de señalización antiviral mitocondrial). LGP2, al carecer del CARD N-terminal, no interactúa con MAVS ni se considera que actúe como molécula de señalización. Durante la infección por el virus sincitial respiratorio (VSR), los macrófagos alveolares producen interferones de tipo I mediante la activación del complejo RIG-I-MAVS, lo que conduce a un mayor reclutamiento de monocitos inflamatorios y limita la replicación viral. Curiosamente, se ha demostrado que el coronavirus del síndrome respiratorio agudo grave (SARS-CoV) y el SARS-CoV-2 pandémico interfieren con la activación de RIG-I para limitar la producción de interferón de tipo I, aumentando así la replicación viral. Trabajos recientes han demostrado que RIG-I limita la replicación del SARS-CoV-2 en células pulmonares mediante un mecanismo no canónico, independiente de MAVS, que depende del dominio helicasa de RIG-I. Si bien su relevancia biológica debe confirmarse in vivo, esto podría representar un mecanismo único para potenciar la inmunidad frente a la infección por coronavirus. Además, la nucleoproteína del SARS-CoV-2 interfiere con la activación de MAVS por RIG-I a través de su dominio helicasa, impidiendo la activación del interferón de tipo I. La interferencia viral fue inhibida por un péptido que actúa sobre el dominio de dimerización de la nucleoproteína viral, previniendo así el daño pulmonar grave y limitando la replicación viral en ratones transgénicos humanizados infectados con SARS-CoV-2. Estos nuevos hallazgos sugieren que la señalización RLR es fundamental para limitar la infección por coronavirus, cuyos mecanismos probablemente también sean relevantes para otros virus de ARN. Finalmente, si bien la defensa del huésped mediada por RLR se ha atribuido en gran medida a la infección viral, estudios han demostrado que Legionella pneumophila, el agente causal de la enfermedad del legionario, puede activar la vía RIG-I/MDA5 debido a su capacidad para inyectar material genético en el citosol mediante un sistema de secreción de tipo IV. Futuros estudios deberían considerar el papel de la señalización RLR en otras especies bacterianas intracelulares.
Nombre |
Ligando |
Ubicación |
Función |
Señalización |
Comentario |
RIG-I |
ARN viral |
Citoplasma |
Producción de interferón tipo I y III |
MAVS |
|
MDA5 |
ARN viral |
Citoplasma |
Producción de interferón tipo I y III |
MAVS |
|
LGP2 |
ARN viral |
Citoplasma |
Efecto antiviral poco claro |
Ninguno |
Posible regulador negativo de RIG-I y MDA5 |
Tabla 3. Receptores tipo RIG-I (RLR)
Receptores sensores de ADN
Además de los receptores de reconocimiento de patrones que reconocen ARN, existen receptores de detección de ADN (Tabla 4), como los receptores tipo AIM2 (similares a los ausentes en el melanoma 2, ALR), la sintasa de GMP-AMP cíclico (cGAS) y la proteína 16 inducible por interferón gamma (IFI16). La mayoría de estos receptores utilizan STING (estimulador de genes de interferón) como proteína adaptadora. La vía de reconocimiento de patrones cGAS-STING es un sistema intracelular de detección de ADN que da como resultado la activación del interferón tipo I. En los pulmones, se ha informado que cGAS reconoce bacterias grampositivas y gramnegativas, como S. pneumoniae y Pseudomonas aeruginosa, y media la protección contra la neumonía causada por la infección por P. aeruginosa. Los ratones STING−/− de cuerpo entero tienen niveles elevados de citocinas inflamatorias, edema alveolar, necroptosis de macrófagos y un aumento de la carga bacteriana durante la infección por S. aureus. La proteína 204 inducible por interferón (IFI204; homólogo murino de IFI16) es un sensor de ADN intracelular similar a AIM2, que facilita las respuestas de interferón tipo I a la infección bacteriana intracelular a través de cGAS, STING y el factor regulador de interferón 3 (IRF3). Más recientemente, se ha demostrado que IFI204 protege contra la infección pulmonar extracelular por S. aureus de forma independiente de la inducción de interferón tipo I. La ablación de IFI204 en ratones provocó una disminución de la supervivencia y un aumento de la carga bacteriana y del daño pulmonar, junto con una reducción de las trampas extracelulares de macrófagos y neutrófilos. Curiosamente, se ha demostrado que los receptores similares a AIM2 no son esenciales para la producción de interferón tipo I, lo que indica que podrían tener una función alternativa.
Nombre |
Ligando |
Ubicación |
Función |
Señalización |
cGAS |
ADN citosólico |
Citoplasma |
Producción de interferón tipo I y citocinas proinflamatorias |
STING |
IFI16 / IFI204 |
ADN intracelular de cadena simple y doble |
Núcleo |
Interferón tipo I |
STING |
Tabla 4. Receptores sensores de ADN
Receptores de lectina de tipo C
Los receptores de lectina de tipo C son una superfamilia heterogénea de receptores, que comprende más de 1000 proteínas, subdivididas en 17 grupos más pequeños. Realizan una amplia variedad de funciones biológicas, que incluyen, entre otras, la regulación de la apoptosis, el crecimiento y el desarrollo, y la inmunidad antimicrobiana. Con respecto a esto último, muchos receptores de lectina de tipo C, como las colectinas y la dectina-1, sirven como receptores de reconocimiento de patrones (Tabla 5). La mayoría de los receptores de lectina de tipo C permanecen sin caracterizar en sus funciones contra patógenos respiratorios, sin embargo, el miembro D de la familia 4 del dominio de lectina de tipo C (CLEC4D), es protector contra la neumonía inducida por K. pneumoniae, donde los ratones knockout de Clec4d exhiben mayor mortalidad, daño pulmonar y UFC bacterianas, lo que puede atribuirse a un defecto en la eliminación de neutrófilos. Dectin-2, que se une al patógeno fúngico Pneumocystis jirovecii, activa la señalización de citocinas proinflamatorias en respuesta a la infección fúngica en el pulmón; Sin embargo, la deficiencia de dectina-2 no parece tener un impacto en la carga fúngica. Entre los receptores de lectina de tipo C que responden a la infección respiratoria, como el versicano y la proteína surfactante D, Mincle (receptor de lectina de tipo C inducible por macrófagos) ha sido ampliamente estudiado y se ha demostrado que tiene un papel destacado contra la neumonía. En respuesta a la infección por K. pneumoniae, la deficiencia de Mincle condujo a un aumento progresivo de la carga bacteriana, a pesar de una respuesta inflamatoria exagerada que promovió un daño pulmonar grave. Además, los ratones con deficiencia de Mincle mostraron defectos en la fagocitosis de neutrófilos y la formación de NET, lo que puede explicar parcialmente su incapacidad para controlar la infección. Mincle también reconoce S. pneumoniae, sin embargo, su papel en la respuesta a la infección neumocócica es inconsistente en la literatura. En un estudio, los ratones knockout de Mincle no pudieron eliminar las bacterias de los pulmones, lo que se acompañó de una respuesta inflamatoria aumentada. Sin embargo, a diferencia de la infección por K. pneumoniae, no mostraron deficiencias en la fagocitosis de neutrófilos. Un segundo estudio demostró que los ratones knockout de Mincle no presentaban cambios en las respuestas inflamatorias, la fagocitosis ni la eliminación bacteriana en respuesta a la infección por S. pneumoniae. Finalmente, un estudio más reciente mostró que la sobreexpresión transgénica de Mincle en ratones conducía a la activación del inflamasoma NLRP3, lo que provocaba un aumento de la lesión pulmonar y la mortalidad en respuesta a la neumonía neumocócica, lo cual podía revertirse mediante la inhibición del inflamasoma NLRP3. Los ratones deficientes en Mincle también presentan una mayor carga fúngica en respuesta a la infección por P. jirovecii, aunque sin cambios en la mortalidad, lo que podría atribuirse a un aumento de la proteína antiinflamatoria IL1RA. En resumen, es probable que muchos más receptores de lectina de tipo C influyan en la infección pulmonar, pero aún no se han explorado. Por ejemplo, se ha demostrado que el receptor de lipoproteína de baja densidad oxidada similar a la lectina-1 (LOX-1) atenúa la lesión pulmonar inducida por sepsis en modelos donde se inhibe sistémicamente, pero aún se desconoce su contribución a la respuesta local en el pulmón. Estos y otros aspectos deberían ser objeto de futuras investigaciones.
Nombre |
Ligando |
Ubicación |
Grupo |
Señalización |
CLEC4D (MCL) |
α-mananos |
Superficie celular |
II – Receptores tipo 2 |
NF-κB |
Proteína surfactante A y D |
Patógenos respiratorios |
Secretado |
III – Colectinas |
Aumento de la fagocitosis y vía de reparación IL4/IL13 |
Dectina-1 |
β-glucano; galectina-9; anexinas; vimentina; tropomiosina; N-glicano |
Membrana celular |
V – Receptores NK |
ROS dependiente de Syk; señalización NFAT y NFκB |
Dectina-2 |
α-mananos |
Membrana celular |
II – Receptores tipo 2 |
Syk, PKCδ y CARD9–Bcl10–Malt1 |
Versicano |
CD44, PSG-1, TLR2; selectinas P y L |
Extracelular |
I – Proteoglucano |
MyD88 |
Mincle |
Glucosil-diacilglicerol (Gly-DAG); especies lipídicas microbianas |
Superficie celular |
II – Receptores tipo 2 |
Syk, PKCδ, CARD9–Bcl10–Malt1 y MAPK |
Tabla 5. Receptores de lectina de tipo C
Receptores depuradores
Los receptores depuradores son un conjunto muy diverso de receptores celulares que se han subdividido en diez clases según sus características estructurales y función biológica (Tabla 6). Una característica común a todos los receptores depuradores es que se unen a un grupo heterogéneo de ligandos, que incluyen, entre otros, lipoproteínas modificadas, componentes microbianos (LPS, ácido lipoteicoico (LTA) y CpG), células apoptóticas y productos finales de glicación avanzada. La proteína C reactiva (PCR) es otro ligando conocido de algunos receptores depuradores, incluidos LOX-1 y SR-A, y desempeña un papel importante en la defensa del huésped contra las bacterias mediante la opsonización mediada por el complemento. La fijación del complemento, un mecanismo altamente conservado de defensa y regulación inmunitaria mediante el reconocimiento de patrones moleculares asociados a microorganismos y daños, es un factor crítico en la defensa del huésped y en la respuesta rápida a la infección bacteriana, cuyos detalles se han revisado en otras publicaciones. Además de la unión a ligandos, se ha demostrado que algunos receptores depuradores, como SR-A, LOX-1, P2X7 y SSc5D, también se unen directamente a las bacterias, lo que contribuye a su captación y eliminación. Los receptores depuradores desempeñan diversas funciones en la salud y la enfermedad, mostrando funciones tanto proinflamatorias como antiinflamatorias dependiendo de la ubicación del receptor, el ligando y la capacidad de interactuar con correceptores, como TLR4. Durante la neumonía, existe una escasez de datos recientes sobre la función de la mayoría de los receptores depuradores, aunque estudios anteriores han mostrado funciones destacadas para MARCO (receptor de macrófagos con estructura colagenosa), CD36 y SR-A (receptor depurador A) en respuesta a la infección pulmonar. Sin embargo, recientemente se demostró que CD5L (molécula similar a CD5), que es un receptor depurador de la superfamilia rica en cisteína, contribuye a una respuesta murina más grave a Staphylococcus aureus resistente a la meticilina (SARM) en el pulmón. La inhibición de CD5L mediada por anticuerpos condujo a una menor mortalidad, junto con una disminución de los niveles de citocinas pulmonares y una menor carga bacteriana, mientras que la suplementación con CD5L aumentó la mortalidad, la lesión pulmonar, las citocinas pulmonares y la carga bacteriana. Curiosamente, la adición de CD5L también aumentó la captación fagocítica de SARM en macrófagos y neutrófilos, pero no tuvo efecto sobre la eliminación bacteriana. Por otro lado, el receptor depurador CD36 promueve la fagocitosis y la eliminación de K. pneumoniae de manera independiente de la cápsula. Los ratones deficientes en CD36 también tienen un perfil de citocinas reducido y una fagocitosis alterada, probablemente como resultado de una menor sensibilidad al LPS. Finalmente, SR-BI (receptor depurador de clase B tipo I), un receptor depurador que media la captación de ésteres de colesterol, también es importante para la defensa pulmonar contra K. pneumoniae. Los ratones deficientes en SR-BI tienen un fenotipo muy similar al de aquellos que carecen de CD36, en el que la mortalidad, la lesión pulmonar, los niveles de citocinas inflamatorias y la replicación y diseminación bacteriana están aumentados. Además, se reclutan más neutrófilos al pulmón de forma dependiente de la corticosterona y, al igual que con los receptores depuradores mencionados anteriormente, la fagocitosis se ve afectada en ratones deficientes en SR-BI. Finalmente, se reveló que SR-BI es necesario para la eliminación eficiente de LPS, sin el cual la señalización de LPS se prolonga. En conjunto, los datos actuales sugieren que los receptores depuradores desempeñan un papel importante en la defensa contra los patógenos pulmonares, y se requiere un trabajo considerable para dilucidar sus funciones específicas, que dependen del microorganismo.
Defensa impulsada por leucocitos
Los leucocitos participan en la respuesta inmunitaria innata actuando mediante funciones centinela y/o efectoras. Las células centinela residen en el tejido local y responden rápidamente a la presencia de PAMP y DAMP en el entorno. Las células efectoras generalmente circulan y son reclutadas al sitio de infección o lesión por señales producidas por las células centinela. Algunas células inmunitarias innatas pueden actuar tanto como células centinela como efectoras, como los macrófagos alveolares y los linfocitos innatos. En esta sección, analizaremos los leucocitos que participan en la respuesta inmunitaria innata.
Macrófagos alveolares
Los macrófagos alveolares (MA) son las células inmunitarias residentes mejor caracterizadas del pulmón. Como su nombre indica, se localizan en el compartimento alveolar, donde constituyen la primera línea de defensa contra los microorganismos invasores gracias a su elevada capacidad fagocítica. Además, promueven la reparación tisular y la homeostasis pulmonar. Los MA son células de larga vida que se encuentran muy cerca de las células epiteliales alveolares de tipo II, donde son responsables de la ingestión y degradación del surfactante pulmonar y de mantener la homeostasis tisular incluso tras una lesión. El GM-CSF (factor estimulante de colonias de granulocitos y macrófagos) derivado del epitelio promueve el establecimiento y la diferenciación de los MA mediante la activación del PPAR-γ (receptor activado por proliferadores de peroxisomas γ), cuya ausencia provoca un desarrollo defectuoso de los MA y proteinosis alveolar pulmonar debido a la acumulación de surfactante. El grupo inicial de macrófagos alveolares (MA) se deriva de precursores del saco vitelino de monocitos fetales, pero se reemplaza gradualmente por macrófagos derivados de monocitos a lo largo de la vida del huésped. La función diferencial de los macrófagos derivados de monocitos fetales frente a los derivados de monocitos es objeto de investigación activa, revisada en. Las intrusiones de bajo nivel en los alvéolos son eliminadas rápida y eficazmente por los MA mediante la producción de proteínas antimicrobianas, especies reactivas de oxígeno y fagocitosis. Una vez que los microorganismos invasores alcanzan un cierto umbral que supera la capacidad de eliminación de los MA, secretan citocinas, quimiocinas y metabolitos del ácido araquidónico para reclutar neutrófilos que ayudarán a eliminar los patógenos. Ambos procesos se analizarán con mayor detalle en las siguientes secciones.
Reconocimiento y eliminación de patógenos
Al igual que la mayoría de las células inmunitarias y no inmunitarias, los macrófagos alveolares (MA) reconocen los microorganismos mediante la expresión de receptores de reconocimiento de patrones (PRR), que se unen a motivos conservados presentes en diferentes clases de microbios. Esta interacción y la señalización subsiguiente alertan al macrófago sobre la amenaza, permitiendo la eliminación directa del microorganismo mediante fagocitosis, óxido nítrico, peróxido de hidrógeno y generación de especies reactivas de oxígeno. Se ha demostrado que el reconocimiento de P. aeruginosa flagelada depende de la expresión de TLR5 en el macrófago, lo que promueve la fagocitosis dependiente de IL-1β y la eliminación del patógeno mediante la producción de asparagina endopeptidasa. Por el contrario, la capacidad fagocítica de los MA se ve disminuida durante la neumonía secundaria debido a un microambiente alterado provocado por una infección primaria con Escherichia coli o S. aureus, lo que conlleva una eliminación deficiente de las bacterias. Aunque aún no se han identificado, los componentes alveolares en ratones recuperados aumentan la expresión de SIRPα (proteína reguladora de la señal α) en los macrófagos alveolares (MA), lo que inhibe su capacidad fagocítica mediante la regulación del metabolismo celular y la transcripción génica. Curiosamente, la eliminación bacteriana mejora tras exposiciones repetidas de ratones a diferentes cepas de S. pneumoniae, lo que conduce a una reprogramación transcripcional y metabólica de los MA. Cabe destacar que, en humanos, la colonización experimental de la nasofaringe con S. pneumoniae (serotipo 6B) aumentó la capacidad opsonofagocítica homo- y heterotípica de los MA. La activación de los MA se correlacionó con la microaspiración de neumococo en el líquido alveolar y dependió de la diferenciación de las células T cooperadoras de tipo 1 (Th1) y los bajos niveles resultantes de IFN-γ, que promovieron la producción de TNF-α por los MA y aumentaron la diferenciación de monocitos a macrófagos. Recientemente se descubrió que las deficiencias en la proliferación de macrófagos alveolares (MA) relacionadas con la edad dependen del microambiente alveolar. El aumento de los niveles de hialuronano en ratones envejecidos confiere un fenotipo hiporreactivo al GM-CSF en los MA, lo que reduce su capacidad proliferativa y aumenta la gravedad de la infección por influenza A. Esto confirmó un informe previo que indicaba que el envejecimiento compromete el número y la función de los MA.
La señalización del receptor de interferón gamma (IFNGR1) en los MA produce resultados contradictorios según la etiología de la infección de las vías respiratorias inferiores. El IFN-γ promueve la eliminación del VRS en ratones neonatales de una manera que depende de los macrófagos pulmonares, y los MA muestran una mayor expresión del complejo mayor de histocompatibilidad II (MHCII), un indicador directo de su activación. Sin embargo, la eliminación bacteriana con una cepa no letal de S. pneumoniae tras la influenza se ve afectada y se vuelve fatal debido a la señalización del IFNGR1 en los MA y otros fagocitos mononucleares. El interferón tipo I exacerba de manera similar la gravedad de la neumonía bacteriana secundaria con SARM, posiblemente a través de la activación del STAT2 (transductor de señal y activador de la transcripción 2) de los macrófagos. La anulación de esta respuesta mejora la eliminación bacteriana debido a la presencia de macrófagos que expresan marcadores de superficie tanto inflamatorios como reguladores (macrófagos M1/M2), que representan macrófagos proinflamatorios frente a macrófagos pro-resolutivos, respectivamente. La infección de ratones con RSV indujo un fenotipo similar a M2 (regulador) en los AM que depende de un aumento en la señalización de Gas6/Axl (proteína 6 específica de detención del crecimiento), típicamente asociada con la eferocitosis. La polarización de los AM resultó en una mayor susceptibilidad a la infección neumocócica secundaria debido a una menor producción de IL-18 por parte de los AM e IFN-γ por parte de las células asesinas naturales. Los estudios futuros deberían centrarse en identificar componentes alveolares críticos y mecanismos de señalización que dan lugar a alteraciones duraderas en la función de los macrófagos alveolares (MA), así como en caracterizar las diferencias específicas de cada patógeno en los cambios fenotípicos mencionados. Si bien los MA residentes suelen ser suficientes para erradicar patógenos menores o menos virulentos y mantener la integridad del tejido alveolar, una mayor carga patógena requiere la ayuda adicional de células inmunitarias reclutadas. Además de la eliminación directa, los MA producen quimiocinas y citocinas, como IL-8, CXCL1, CXCL2 (ligando 1 y 2 de quimiocina con motivo C-X-C), IL-6 y TNF-α, para activar y reclutar células inmunitarias innatas y adaptativas adicionales.
Funciones en el reclutamiento y activación de neutrófilos.
Si bien los macrófagos alveolares (MA) activan tanto la respuesta inmunitaria innata como la adaptativa, el presente estudio se centra en las respuestas agudas iniciales a la infección y, por lo tanto, en el reclutamiento de células derivadas de células progenitoras mieloides. Como se mencionó anteriormente, las agresiones inflamatorias pueden provocar disfunción de los MA y defectos posteriores en el reclutamiento de neutrófilos y un aumento de la carga bacteriana durante la infección secundaria. De hecho, los patógenos que producen toxinas formadoras de poros pueden atacar directamente a los MA, lo que subraya su papel central en la modulación de las respuestas posteriores. Tras la sepsis por ligadura y punción cecal, los MA reducen su producción de CXCL1 y la fagocitosis de bacterias de forma mediada por TNF-α/interleucina-10 (IL-10). Sin embargo, este defecto puede revertirse con la administración de interferón-β (IFN-β), cuya producción puede verse atenuada por el aumento de los niveles del factor de crecimiento transformante-β1 (TGF-β1) tras la lesión pulmonar inducida por la sepsis. La producción de CXCL1 y el reclutamiento temprano de neutrófilos también se ven afectados en ratones que carecen de FABP4 (proteína de unión a ácidos grasos-4) de macrófagos, lo que conduce a una mayor replicación bacteriana y mortalidad tras la infección por P. aeruginosa. Curiosamente, la expresión de TLR3 en los AM es perjudicial para la respuesta del huésped a K. pneumoniae, por lo que los macrófagos TLR3−/− exhiben una mayor capacidad fagocítica y eferocítica junto con un fenotipo más similar a M2. La capacidad de respuesta de los macrófagos TLR3−/− también se ve mejorada en que expresan niveles más altos de CXCL1 y CXCL2, lo que conduce a un mayor número de neutrófilos, pero curiosamente exhiben una lesión pulmonar disminuida. Este fenotipo probablemente se deba a los efectos concordantes de la carga bacteriana reducida y la mayor eferocitosis de neutrófilos moribundos [36]. Durante la infección viral, los AM producen niveles robustos de IL-1β a través de la activación del inflamasoma NLRP3. Curiosamente, la activación del inflamasoma NLRP3, pero no su expresión, depende del péptido antimicrobiano relacionado con la catelicidina (mCRAMP), derivado de los neutrófilos. Por lo tanto, durante la infección viral, pero no la bacteriana, la secreción de IL-1β requiere un sistema de doble impacto en los macrófagos alveolares (MA) mediante la infección inicial y la interacción con los neutrófilos reclutados. La producción de IL-1β durante la coinfección por influenza/neumococo previene la apoptosis de los MA, un proceso que protege contra el crecimiento excesivo del serotipo 14 de S. pneumoniae, pero no del 19. Curiosamente, durante la coinfección en ratones deficientes en el receptor de IL-1 tipo 1 (ratones Il1r1−/−), la falta de señalización de IL-1β se compensa con la activación de los neutrófilos reclutados por TNF-α. El TNF-α, producido por los neutrófilos, protege de manera similar contra la infección por L. pneumophila mediante su acción sobre los macrófagos alveolares (MA), donde la señalización del TNF-α aumenta la acidificación lisosomal de forma dependiente de caspasas. Los MA moderan la actividad de los neutrófilos durante la infección por Acinetobacter baumannii, donde tanto el reclutamiento de neutrófilos como la producción de especies reactivas de oxígeno por parte de estos se ven potenciados con la depleción de macrófagos. Esto resulta perjudicial para el huésped infectado por A. baumannii, quien presenta un daño pulmonar y una carga bacteriana notablemente mayores. En resumen, los MA son determinantes cruciales en el reclutamiento de neutrófilos durante la infección del compartimento alveolar, cuyo resultado puede mejorar o empeorar el pronóstico según el origen de la infección.
Mecanismos alternativos de respuesta de los macrófagos alveolares a la neumonía
Además de la eliminación directa y el reclutamiento de leucocitos en los pulmones, los macrófagos alveolares (MA) han desarrollado métodos alternativos para contribuir a la defensa del huésped y la recuperación de la neumonía. Un mecanismo es la apoptosis del propio macrófago para prevenir la replicación microbiana, la cual se ha observado tras la infección por influenza, neumococo y tuberculosis, entre otros patógenos importantes. Recientemente se demostró que esta respuesta se ve inhibida por la sobreexpresión en macrófagos de la proteína antiapoptótica MCL-1 (leucemia de células mieloides 1), lo que dificulta la eliminación de S. pneumoniae y Haemophilus influenzae. Es importante destacar que no se observó apoptosis de macrófagos tras la infección por S. aureus, lo que indica importantes diferencias específicas de cada patógeno en la respuesta celular a la infección.
Finalmente, los MA son fundamentales para la reparación tisular tras la lesión pulmonar inducida por neumonía. Si bien este tema escapa al alcance de esta revisión, cabe mencionar algunos estudios que abren nuevas vías de investigación. La producción del factor de crecimiento resolutivo tisular, anfirregulina, es inducida por los macrófagos alveolares (MA) durante la infección por Nippostrongylus brasiliensis y promueve la restauración de la barrera hemato-pulmonar. Esto ocurre a través de la activación «de adentro hacia afuera» de la integrina-αV en los pericitos, lo que conduce a la liberación de TGF-β y su diferenciación en miofibroblastos. Las propiedades reparadoras de los MA también están reguladas metabólicamente. La activación de la vía de señalización Wnt-β-catenina resulta en un aumento de la expresión del factor inducible por hipoxia 1α (HIF-1α) en macrófagos y un metabolismo glucolítico mayor que el mitocondrial. El desequilibrio metabólico hacia la glucólisis provocó una señalización inflamatoria elevada durante la infección por influenza a expensas de la proliferación y la reparación.
Macrófagos intersticiales
Mientras que los macrófagos alveolares (MA) residen en la luz alveolar, en una proporción aproximada de un macrófago por cada tres alvéolos, los macrófagos intersticiales (MI) habitan el espacio intersticial entre el epitelio y los capilares subyacentes. Se creía que residían exclusivamente cerca de los bronquios; sin embargo, informes recientes han demostrado la existencia de una población de MI CD206− ubicada dentro del intersticio alveolar, y otros han demostrado que pueden atravesar el espacio luminal cuando es necesario. Los MI se han clasificado en tres subtipos distintos (MI1-3) según la expresión de varios marcadores, incluidos CD11c, MHCII, CD206, CD169 y Lyve-1, aunque no está claro si estos representan tres subpoblaciones fenotípicamente distintas. Dos estudios recientes han identificado dos grupos únicos de IM basados en la expresión de CD206 o Lyve1/MHCII/CX3CR1 y una población que puede representar un estado de transición de monocitos patrulladores Ly6c-bajos (CD64+CD16.2+). Otro estudio identificó una población de IM CD169+ derivados de embriones, asociados a nervios, que pueden ser los mismos que la población Lyve1bajo/MHCIIalto identificada en el trabajo de Chakarov et al. Ontológicamente, los IM tienen un trasfondo mixto, originándose tanto de monocitos residentes en sangre y pulmón como de precursores del saco vitelino, y esta variación puede contribuir a su heterogeneidad. El papel de los IM en respuesta a la neumonía está menos caracterizado que el de los AM; sin embargo, los IM exhiben profundas propiedades inmunorreguladoras en su capacidad para producir IL-10. En respuesta a múltiples ligandos, pero especialmente al ADN CpG, los IM se expanden y producen grandes cantidades de IL-10, que protege contra la enfermedad alérgica de las vías respiratorias. La producción de IL-10 se ha atribuido a la población de IM CD206+ asociada a los bronquios, junto con otras citocinas inmunorreguladoras, como el antagonista del receptor de interleucina-1 (IL1-Ra) y el factor inhibidor de la leucemia (LIF). Sin embargo, las IM CD169+ asociadas a los nervios también producen cantidades sustanciales de IL-10 en respuesta a la infección por influenza y a la estimulación con poli(I:C). La depleción de este subconjunto de IM provoca una mortalidad profunda en ratones infectados con influenza y mayores niveles de citocinas inflamatorias en respuesta al poli(I:C). Además, las señales producidas por las células endoteliales desempeñan un papel importante en la expansión y regulación de las IM. Recientemente se ha demostrado que la respondina-3, un ligando WNT, es producida por las células endoteliales para promover el fenotipo antiinflamatorio de las IM mediante modulación metabólica y epigenética. Gracias a estos cambios, los ratones fueron menos susceptibles a la lesión pulmonar inducida por LPS. Cabe destacar que, si bien los macrófagos alveolares (MA) se infectan preferentemente con Mycobacterium tuberculosis y constituyen un medio para la diseminación microbiana, los macrófagos intestinales (MI) son más capaces de limitar el crecimiento bacteriano mediante mecanismos que dependen de la activación del factor nuclear κB (NF-κB) y del metabolismo glucolítico.
Monocitos y macrófagos derivados de monocitos
Los monocitos son un tipo de célula reclutada que se produce continuamente a lo largo de la vida a partir de células progenitoras hematopoyéticas en la médula ósea. Son una población heterogénea de células que pueden dar lugar a macrófagos derivados de monocitos o células dendríticas derivadas de monocitos. Además, se han identificado monocitos clásicos (Ly6Chi) y monocitos patrulladores (Ly6clo) en ratones y humanos. Se sigue identificando una mayor heterogeneidad entre las poblaciones de monocitos mediante el uso de técnicas de célula única, como la secuenciación de ARN de célula única, aunque sus diferencias funcionales aún no se han determinado [150]. Los receptores de quimiocinas, como el CCR2 (receptor de quimiocina con motivo C-C 2), desempeñan un papel crítico en el reclutamiento de monocitos en el pulmón durante la infección. Por lo tanto, el agotamiento de los monocitos CCR2+ provocó una mayor pérdida de peso y carga bacteriana en los pulmones de ratones infectados con las cinco cepas de K. pneumoniae que se analizaron. Se demostró además que la eliminación inflamatoria de K. pneumoniae por monocitos depende de su producción de TNF-α, que activa los linfocitos innatos tipo 3 (ILC3) para producir IL-17A, promoviendo la producción de ROS por los monocitos. La deficiencia dependiente de la edad en la defensa del huésped contra S. pneumoniae también se ve afectada por los monocitos Ly6Chi, donde los niveles continuos de TNF-α en la sangre de ratones viejos promueven la movilización de monocitos inmaduros, CCR2+ que son menos eficientes en la eliminación bacteriana, enfatizando la importancia de los monocitos completamente funcionales en la defensa del huésped. Recientemente, ha habido un aumento sustancial en el estudio del papel de los monocitos en la promoción de la lesión pulmonar durante la neumonía, debido a su papel implícito en la infección por SARS-CoV-2. Si bien se ha revisado en gran detalle en otras publicaciones, existe evidencia considerable que sugiere que los monocitos (y los macrófagos) contribuyen a la inmunopatología en la infección grave por SARS-CoV-2. De manera similar, los monocitos de pacientes con sepsis y síndrome de dificultad respiratoria aguda (SDRA), en comparación con los pacientes con sepsis únicamente, revelaron una firma genética enriquecida con genes estimulados por IFN y una regulación a la baja de genes de inmunomodulación, como SOCS3 (supresor de la señalización de citoquinas 3).
Neutrófilos
Los neutrófilos son células efectoras altamente especializadas y de corta duración del linaje mieloide; son las primeras células circulantes que se reclutan en el sitio de infección en la neumonía y desempeñan un papel importante en la eliminación de patógenos invasores. Durante las infecciones agudas, los neutrófilos funcionan para eliminar bacterias patógenas a través de tres procesos principales: fagocitosis, degranulación y NETosis. Estos procesos se han revisado ampliamente en otras publicaciones y se resumen brevemente aquí. Al llegar al sitio de infección, los neutrófilos identifican los patógenos uniendo componentes microbianos a PRR, o microbios opsonizados a receptores Fc (FcR) o receptores del complemento (CR) de los neutrófilos. Los microbios unidos son fagocitados, y los fagosomas que contienen bacterias se fusionan con vesículas lisosomales especializadas que contienen, creando un fagolisosoma en el que las bacterias son eliminadas a través de vías oxidativas y no oxidativas. Los microbios que permanecen fuera del neutrófilo también pueden ser eliminados mediante la degranulación de este último, proceso en el que los gránulos preempaquetados se fusionan a la superficie del neutrófilo, liberando su contenido tóxico (proteasas y especies reactivas de oxígeno, ROS) al espacio circundante. Finalmente, mediante la condensación coordinada de la cromatina, la unión de proteínas citoplasmáticas y granulares, y la extrusión del complejo ADN-proteína, los neutrófilos pueden liberar trampas extracelulares de neutrófilos (NET), que transforman el entorno extracelular, atrapando y eliminando los microbios cercanos. Si bien son cruciales para la eliminación de patógenos, estas funciones de los neutrófilos suelen ser la causa de la importante muerte celular y el daño tisular observados en la neumonía y el síndrome de dificultad respiratoria aguda (SDRA). Los neutrófilos también influyen directamente en la respuesta inmunológica de otras células mediante la alteración de marcadores de superficie, la producción de citocinas y el contacto celular directo. Por último, los neutrófilos presentan una dinámica transcripcional, regulando las vías inflamatorias a nivel transcripcional en respuesta a diversos estímulos.
Si bien muchos aspectos de la biología de los neutrófilos son aplicables a todos los sistemas orgánicos, la estructura pulmonar tiene un impacto directo en ella. El lecho capilar pulmonar está compuesto por segmentos estrechos, con un diámetro vascular que oscila entre 2 y 15 µm, en comparación con el diámetro promedio de los neutrófilos (6-8 µm), y presenta una presión menor que la de la circulación sistémica. Debido a este sistema estrecho y de baja presión, los neutrófilos transitan lentamente por el lecho capilar pulmonar, lo que da lugar a una gran cantidad o «reservorio marginal» de neutrófilos en los pulmones en un momento dado. En términos de reclutamiento de neutrófilos, el lento tiempo de tránsito a través del pulmón elimina la necesidad de la captura y el rodamiento de neutrófilos mediados por selectinas e integrinas, procesos fundamentales para la migración de neutrófilos fuera de la vasculatura en otros órganos. La señalización mediada por selectinas e integrinas no es necesaria para la migración de neutrófilos fuera de la vasculatura en la neumonía por S. pneumoniae, pero sí lo es en el caso de otros patógenos. Además, los receptores de adhesión no canónicos también participan en el reclutamiento y la migración de neutrófilos en infecciones respiratorias.
La evidencia emergente sugiere que las respuestas de los neutrófilos, particularmente en lo que respecta a la producción de citocinas y la expresión de marcadores de superficie, pueden variar según la localización tisular y/o el estímulo patógeno. Los neutrófilos que han migrado a los pulmones presentan un transcriptoma distinto al de los neutrófilos circulantes tras la inhalación de endotoxinas o una infección grave por VRS. Asimismo, el repertorio de quimiocinas secretadas por los neutrófilos pulmonares difiere del de los neutrófilos circulantes en la gripe. La caracterización completa de las respuestas de los neutrófilos pulmonares durante la infección está en curso.
Además de manifestar respuestas específicas de cada tejido, también parece que los neutrófilos en cada tejido pueden estar compuestos por subpoblaciones funcionalmente distintas. Estudios recientes que emplean citometría de flujo y secuenciación de células individuales han demostrado la existencia de subpoblaciones de neutrófilos que pueden mantenerse tanto en condiciones basales como durante la infección. Esta heterogeneidad de los neutrófilos se ha caracterizado de diversas maneras, como N1 frente a N2, alta densidad frente a baja densidad, activados frente a refractarios, o inflamatorios frente a inhibidores. Si bien existen pruebas sólidas de la heterogeneidad de las respuestas de los neutrófilos, aún no está claro cuán universal es esta heterogeneidad, ni si las subpoblaciones de neutrófilos se reproducen en el pulmón durante la neumonía.
Linfocitos innatos
Además de su conocido papel en la inmunidad adaptativa, un subconjunto de linfocitos también desempeña un papel importante en la inmunidad innata. Estas células se caracterizan por tener pocos o ningún receptor de antígeno y responden, en cambio, a las señales de alarma del huésped y a los productos de los patógenos. Responden rápidamente a los estímulos y desempeñan un papel crucial al interactuar con otras células efectoras innatas (como macrófagos y neutrófilos), así como con las respuestas inmunitarias adaptativas mediadas por linfocitos T, linfocitos B y células dendríticas. Estos linfocitos innatos se dividen en linfocitos T con diversidad antigénica limitada (linfocitos T γδ, células iNKT y células MAIT) y células linfoides que carecen de receptores de antígeno, pero con morfología linfocitaria (células linfoides innatas: células NK, ILC1, ILC2, ILC3 y células LTi). En esta sección, nos centraremos en aquellos linfocitos innatos con un papel claro en la inmunología pulmonar, específicamente los linfocitos T γδ, las células iNKT, las células MAIT, las células NK y las células ILC2.
Células T innatas
Células T γδ Estas células expresan heterodímeros del receptor de células T (TCR) que contienen cadenas gamma y delta, con una diversidad limitada, y no expresan CD4 ni CD8. En la neumonía, la función principal de las células T γδ es la respuesta temprana a los patógenos. Múltiples señales estimulan estas células, incluyendo la unión directa del antígeno al TCR, la unión de PAMP y DAMP a los TLR o la estimulación directa de citocinas. Las células T γδ activadas producen IL-17A e IFNγ, lo que promueve el reclutamiento de células efectoras (macrófagos y neutrófilos), la formación de granulomas y las respuestas inmunitarias Th17. Múltiples patógenos pulmonares inducen el número de células T γδ mediante la expansión celular. La función específica de las células T γδ en la neumonía puede depender del patógeno, ya que la depleción de estas células resulta en una menor producción de IFNγ y la incapacidad de eliminar el patógeno en infecciones por K. pneumoniae y S. pneumoniae. Por el contrario, el agotamiento de las células T γδ mejoró la eliminación de patógenos y aumentó el IFNγ en un modelo de criptococo [205] y aumentó la producción de IL-17 y la protección contra la infección mortal en un modelo de influenza.
Célula T asesina natural invariante (iNKT) Las células NKT invariantes (iNKT, también conocidas como células NKT clásicas o de tipo I) expresan un TCR αβ con una cadena α invariante y una de las tres cadenas β [209]. Estas células responden a antígenos lipídicos presentados por la molécula CD1d, similar a la del complejo mayor de histocompatibilidad de clase I (MHC). Estos antígenos incluyen la alfa-galactosilceramida (α-Gal-Cer) y diversos lípidos derivados de microorganismos y del huésped, lo que les permite responder a una amplia gama de patógenos. Cabe destacar que los tetrámeros de CD1d cargados con α-Gal-Cer son una herramienta útil para estudiar las células NKT humanas y de ratón. Las células NKT también responden directamente a la estimulación por las citocinas IFNβ, IL-1β, IL-12, IL-18 e IL-23.
Las subpoblaciones de células NKT (NKT1, NKT2 y NKT17) se definen por sus programas de desarrollo y el repertorio de citocinas que expresan. Los pulmones presentan una alta concentración de células NKT17 en el parénquima pulmonar, con un menor número de células NKT1 y NKT2 en la zona vascular marginal. Las células NKT1 producen IFNγ y expresan el factor de transcripción T-box 21 (T-bet). Las células NKT2 producen interleucina-4 (IL-4), expresan PLZF (proteína de dedo de zinc de la leucemia promielocítica) y dependen de PLZF, la proteína de unión a GATA 3 (GATA3) y el factor regulador del interferón 4 (IRF4) para su desarrollo. Finalmente, las células NKT17 producen IL-17 y expresan RORγt (receptor huérfano γ relacionado con el receptor de ácido retinoico).
Las células NKT desempeñan un papel fundamental en la defensa contra múltiples patógenos pulmonares. Son esenciales para la eliminación de bacterias respiratorias como S. pneumoniae, P. aeruginosa, M. tuberculosis y Chlamydia pneumoniae, mediante el reclutamiento de células efectoras como neutrófilos y macrófagos alveolares a través de la producción de quimiocinas y citocinas. Las células NKT también protegen contra patógenos virales como la gripe. Las células dendríticas infectadas con influenza A estimulan las células NKT, lo que resulta en una rápida producción de interleucina-22 (IL-22) y modifica la actividad de las células supresoras derivadas de mieloides y los monocitos proinflamatorios.
Células T invariantes asociadas a la mucosa (MAIT) Al igual que las células NKT, las células T invariantes asociadas a la mucosa (células MAIT) también poseen un TCR restringido, pero responden al receptor MR1 (relacionado con el complejo mayor de histocompatibilidad de clase I), en lugar de CD1d. El MR1 presenta metabolitos de riboflavina (vitamina B2) producidos por diversas especies bacterianas y de levaduras. Además, las células MAIT pueden ser estimuladas por vías independientes del TCR, incluida la señalización de IL-18. Las células MAIT también se pueden subdividir según las citocinas que producen. Hasta el momento, se han identificado MAIT1 (productoras de IFNγ) y MAIT17 (productoras de IL-17 e IL-22), pero no MAIT2. Las células MAIT muestran heterogeneidad en su respuesta a los microbios y podrían funcionar para distinguir entre patógenos y comensales. Asimismo, el número de células MAIT se reduce en ratones libres de gérmenes, a diferencia de las células iNKT, que aumentan en estos ratones. En ausencia de infección, el número de células MAIT en los pulmones de los ratones es bajo. Sin embargo, este número aumenta drásticamente con la infección por las bacterias intracelulares Salmonella typhimurium, Francisella tularensis y L. pneumophila. Las células MAIT podrían desempeñar un papel protector en la infección, ya que su número se correlaciona negativamente con la gravedad de la enfermedad y la infección por P. aeruginosa en la fibrosis quística, y los ratones MR1−/− son más susceptibles a E. coli y Mycobacterium abscessus. Finalmente, los humanos recuperados de influenza grave presentaban un mayor número de células MAIT en sangre periférica que aquellos que fallecieron.
Células linfoides innatas (ILC)
Las células linfoides innatas (ILC) se definen por una morfología linfoide, pero carecen del receptor de antígeno dependiente de RAG o de marcadores de superficie celular asociados a otros linajes linfoides y mieloides. Al igual que otros linfocitos de tipo innato mencionados, los estudios de parabiosis han demostrado que las ILC residen principalmente en los tejidos y no recirculan. En lugar de responder a antígenos, responden a estímulos directos mediante señales de peligro y estrés. Las primeras células de este tipo en ser identificadas fueron las células asesinas naturales (NK) y las células inductoras de tejido linfoide (LTi). Posteriormente se han identificado otros tipos de células linfoides innatas y las ILC se clasifican actualmente según el conjunto de citocinas que producen. Las ILC1 producen citocinas de tipo I (IFNγ y TNF), las ILC2 producen citocinas de tipo II (IL-4, IL-5, IL-9 e IL-13) y las ILC3 producen IL-17 e IL-22. En el pulmón, la gran mayoría de las ILC son células NK e ILC2, que se analizan con más detalle a continuación.
Células asesinas naturales (NK) Las células asesinas naturales (NK) se identifican por los marcadores de superficie CD127−NKp46+Eomes+ y requieren el factor de transcripción T-bet. Expresan una amplia variedad de receptores de superficie activadores e inhibidores que permiten modular sus respuestas según la combinación de ligandos de unión. En presencia de señales de activación, las células NK pueden eliminar directamente las células diana mediante la liberación de perforina y granzima, reclutar células efectoras adicionales mediante la secreción de citocinas y quimiocinas, o inducir la apoptosis de las células diana mediante la expresión de ligandos inductores de apoptosis como el ligando Fas (FasL) o TRAIL (ligando inductor de apoptosis relacionado con el TNF). Por otro lado, las células NK también pueden producir citocinas antiinflamatorias. En el pulmón, donde una actividad excesiva de las células NK podría ser particularmente perjudicial, estas células presentan una mayor proporción de receptores inhibidores que activadores y una activación estrictamente regulada. Sin embargo, las células NK son importantes durante la infección por K. pneumoniae, donde limitan la diseminación bacteriana y mejoran la supervivencia, en parte debido a su producción de IL-22.
ILC2 Las ILC2 se definen por los marcadores CD90+CD127+RORγt−GATA3hi. Se caracterizan como ILC de tipo cooperador y participan en la defensa contra infecciones por helmintos, además de mantener la integridad de la barrera pulmonar. Se activan en respuesta a IL-1β, prostaglandina D2, interleucina-22 (IL-22), interleucina-25 (IL-25) y linfopoyetina estromal tímica (TSLP). Una vez activadas, regulan positivamente GATA3 para producir interleucina-5 (IL-5) e interleucina-13 (IL-13). En los pulmones, su capacidad para producir grandes cantidades de citocinas de tipo II les confiere un papel importante en la respuesta a infecciones virales y por helmintos, así como en la patogenia del asma alérgica.
Defensa de las células estromales
Se ha demostrado que las células del parénquima pulmonar participan activamente en las respuestas inmunitarias. Estas células actúan como centinelas, produciendo citocinas y quimiocinas en respuesta a los PAMP y liberando DAMP cuando se lesionan, lo que conduce al reclutamiento y la activación de las respuestas de las células efectoras. Durante una infección, las células parenquimáticas producen proteínas antimicrobianas que eliminan directamente a los patógenos y pueden modular las respuestas de los leucocitos residentes y reclutados mediante la expresión de quimiocinas, citocinas y receptores de superficie. Finalmente, la estructura única del pulmón y sus células parenquimáticas subyacen a las respuestas inmunitarias específicas de este órgano.
Células epiteliales
Además de proporcionar una barrera física hermética, las células epiteliales pulmonares, tanto en las vías respiratorias superiores como en los alvéolos, desempeñan un papel activo en la defensa inicial contra los patógenos. Las vías respiratorias están formadas por una población heterogénea de células epiteliales con características únicas, que conforman una capa epitelial pseudoestratificada. El pulmón distal consta de dos tipos principales de células epiteliales: las células epiteliales alveolares de tipo I y tipo II (ATI y ATII, respectivamente). La fina capa única de células ATI es la principal responsable del intercambio de gases, mientras que las células ATII proporcionan una reserva regenerativa para las células ATI y secretan surfactante pulmonar. Recientemente se han revisado los avances en nuestra comprensión de la modulación inmunitaria por parte de las células epiteliales pulmonares en la era de la transcriptómica unicelular.
Células epiteliales de las vías respiratorias
El epitelio de las vías respiratorias superiores está compuesto principalmente por células multiciliadas, secretoras y basales, con subpoblaciones menos frecuentes, como ionocitos, células neuroendocrinas, células en penacho y células caliciformes (raras en ratones, comunes en humanos), intercaladas entre sí. La acción cooperativa de las células ciliadas y secretoras constituye la base del mecanismo de eliminación mucociliar descrito en la sección anterior. Debido a su ubicación y a su importante función antimicrobiana, las células ciliadas suelen ser el objetivo principal de los microorganismos invasores, interfiriendo directa o indirectamente con la eliminación mucociliar. Más recientemente, se ha demostrado que los cilios poseen capacidades de quimiosensación y transducción de señales para potenciar las defensas inmunitarias innatas contra los patógenos, independientemente de la eliminación directa. En 2009, se descubrió que los receptores del gusto amargo (T2R) se expresan en los cilios y que, al activarse, aumentan la frecuencia del batido ciliar de forma dependiente del calcio. Curiosamente, los ligandos T2R, la acil-homoserina lactona y las quinolonas, son secretados por P. aeruginosa como moléculas de detección de quórum, lo que resulta en una mayor producción de óxido nítrico en cultivos de interfase aire-líquido. En conjunto, estos resultados sugieren que las células ciliadas pueden detectar estas moléculas y potenciar la actividad inmunitaria y el batido ciliar en respuesta a la infección. Por el contrario, la activación no canónica de la señalización Hedgehog reduce los niveles de AMPc en las células ciliadas, lo que resulta en una menor frecuencia de batido ciliar. Las células multiciliadas de las vías respiratorias superiores también se pueden distinguir por la expresión de una proteína similar a Piwi, MIWI2, cuya ausencia resulta en una mejor eliminación bacteriana durante la neumonía neumocócica debido a una mayor expresión de proteínas inflamatorias y al reclutamiento de células inmunitarias.
Células epiteliales alveolares
Los alvéolos distales están compuestos por células epiteliales alveolares tipo I y tipo II (ATI y ATII). Las ATI son células epiteliales planas y escamosas que constituyen el principal lugar de intercambio gaseoso en el pulmón. Las células ATII son células epiteliales cúbicas que producen surfactante pulmonar y otras proteínas antimicrobianas, descritas previamente. Es importante destacar que los neumocitos ATII también constituyen una reserva de células progenitoras, ya que tienen la capacidad de diferenciarse en células tipo I si sufren una lesión. Este proceso está regulado por la señalización Wnt en las células tipo II Axin+, lo que impide su diferenciación en condiciones homeostáticas y potencia la proliferación de ATII tras una lesión pulmonar inducida por la gripe. Por otro lado, la señalización BMP (proteína morfogenética ósea) en las células ATII promueve la diferenciación a células tipo I, siendo los fibroblastos PDGFR-α (receptor α del factor de crecimiento derivado de plaquetas) la principal fuente de ligandos tanto de Wnt como de BMP.
Si bien existe evidencia considerable de la capacidad de las células ATII para responder a la infección pulmonar, como lo demuestra la reducción de la eliminación bacteriana en ratones que expresan una forma dominante negativa de IκB-α en células tipo II, se sabe mucho menos sobre la capacidad de respuesta de las células ATI. Sin embargo, en respuesta a la neumonía neumocócica, se ha demostrado que las células ATI producen CXCL5 (ligando 5 de quimiocina con motivo C-X-C) de manera independiente de RelA. Recientemente, se demostró que la expresión de EMP2 (proteína de membrana epitelial 2) en células ATI promueve la afluencia de neutrófilos tras la infección por K. pneumoniae. En este caso, el aumento de los niveles de neutrófilos empeoró el pronóstico de la neumonía y dependió de las propiedades de unión al colesterol de EMP2, que facilitaron la formación de balsas lipídicas, un factor conocido que mejora la señalización de PRR. Estos estudios implican la importancia de las células ATI en la respuesta temprana a la infección, aunque aún quedan muchas preguntas sin respuesta. Su fragilidad hace que sea especialmente difícil trabajar con ellas, lo que requiere técnicas mejoradas para el aislamiento celular o análisis in vivo para evaluar a fondo el papel de las células ATI durante la neumonía.
Eliminación directa Las células epiteliales de las vías respiratorias producen la potente antimicrobiana lisozima, que hidroliza directamente los componentes de la pared celular bacteriana, y la lipocalina-2, un sideróforo que limita el crecimiento de K. pneumoniae y E. coli. Como muestra de la importancia de la regulación del hierro durante la neumonía, recientemente se demostró que la hepcidina, derivada del epitelio de las vías respiratorias, limita los niveles de hierro en los pulmones al actuar sobre la ferroportina, el transportador de hierro. Los ratones con deficiencia de hepcidina presentaron una neumonía más grave debido al crecimiento bacteriano excesivo y a la deficiencia en la función de los macrófagos. La LL-37, la subunidad C-terminal activa de la catelicidina humana, también es producida por las células epiteliales de las vías respiratorias y las glándulas submucosas, contribuyendo directamente a la muerte celular bacteriana mediante interacciones con la membrana. La LL-37 también promueve la activación inflamatoria al actuar como quimioatrayente y la modulación inmunitaria mediante el secuestro de moléculas con carga negativa, como el LPS y diversas DAMP. Las células epiteliales también pueden sintetizar componentes de la vía del complemento que ayudan a la defensa contra patógenos bacterianos, virales y fúngicos. Las células epiteliales respiratorias infectadas por el SARS-CoV-2 expresan múltiples proteínas del complemento de forma dependiente de JAK 1/2 (Janus quinasa 1/2), incluyendo una C3 convertasa funcional. Si bien el complemento es beneficioso en respuesta a diversas infecciones pulmonares, su activación excesiva se ha asociado con una mayor gravedad de la infección por SARS-CoV-2.
Funciones en el reclutamiento de leucocitos Además de la inhibición microbiana directa, las células epiteliales también promueven el reclutamiento de leucocitos, principalmente mediante la producción directa o indirecta de quimiocinas y citocinas. Tras una neumonía neumocócica, las células epiteliales de las vías respiratorias conductoras producen SECTM1 (proteína 1 secretada y transmembrana), que se une selectivamente a los neutrófilos en el pulmón infectado, induciéndolos a producir CXCL2 y generando un ciclo de retroalimentación positiva para el reclutamiento de neutrófilos. Además, con la exposición repetida al neumococo, las células epiteliales se reprograman para producir niveles elevados de CXCL5 de forma dependiente de RelA y de las células T, lo que resulta en un mayor reclutamiento de neutrófilos en las primeras etapas posteriores a la infección y una eliminación bacteriana más eficaz en comparación con animales no expuestos. La señalización de RelA en las células epiteliales también promueve la expresión de CCL20 (ligando 20 del motivo C-C) y GM-CSF; este último ofrece una protección significativa contra la neumonía grave, probablemente debido a sus múltiples efectos sobre los macrófagos alveolares vecinos. El GM-CSF también protege contra la infección por M. tuberculosis, debido a la disminución de los niveles de formación de NET de neutrófilos dependiente de interferón tipo I. Además, los macrófagos infectados por L. pneumophila producen IL-1 que estimula la producción de GM-CSF en las células epiteliales alveolares, lo que conduce a la activación de monocitos reclutados a través de un aumento de la glucólisis y la producción de citocinas, cuyas acciones coordinadas conducen a un mejor control de la infección. Las células epiteliales también expresan IL-17R y son altamente sensibles a múltiples miembros de la familia IL-17. Después de la infección por K. pneumoniae, la ablación de IL-17RA (receptor A de interleucina-17) en células club que expresan SCGB1A1 (miembro 1 de la familia 1A de la secretoglobina) reduce significativamente los niveles de CXCL5, lo que conduce a una menor captación de neutrófilos y una mayor carga bacteriana. La oncostatina-M (OSM), una citocina de la familia IL-6, también aumenta la expresión de CXCL5 y la afluencia de neutrófilos a los pulmones tras la infección por E. coli, probablemente como resultado de la activación específica de STAT3 (transducción de señales y activador de la transcripción 3) por parte de OSM en las células epiteliales pulmonares.
Reparación y recuperación Las células epiteliales también poseen importantes propiedades regenerativas, esenciales para la recuperación tras una infección. Si bien estas propiedades regenerativas no son aspectos de la inmunidad innata propiamente dicha, las señales y los programas inmunitarios innatos pueden afectar la regeneración. Por ejemplo, tras una infección por influenza, la IL-33 derivada del epitelio promueve la producción de anfirregulina en las células linfoides innatas (ILC) del pulmón, facilitando la reparación. En contraste, los interferones de tipo I (IFN-α e IFN-β) y de tipo III (IFN-λ), inducidos tras la infección por influenza, interfieren con la reparación y diferenciación epitelial. La señalización de interferón en las células epiteliales conduce a la activación de p53, un mediador clave de la detención del ciclo celular y la apoptosis, lo que empeora el pronóstico ante una infección secundaria por S. pneumoniae. Se observaron resultados similares durante la infección por K. pneumoniae, donde la integridad epitelial se vio afectada por el IFN-λ y protegida por la expresión de IL-22. Los coactivadores transcripcionales YAP (proteína 1 asociada a Yes) y TAZ (coactivador transcripcional con motivo de unión a PDZ) son reguladores negativos de la vía Hippo, una vía conservada evolutivamente que regula el desarrollo y la regeneración tisular. Recientemente, se investigó la activación de YAP/TAZ en el contexto de la regeneración alveolar tras la infección neumocócica, que afecta principalmente a los alvéolos pulmonares. En este estudio, los autores demuestran que la translocación nuclear de YAP/TAZ inducida por la neumonía promueve la diferenciación de ATII a ATI entre los 7 y 14 días posteriores a la infección, cuya ausencia provocó lesiones fibróticas en el pulmón y una recuperación más lenta. La fibrosis pulmonar se atribuyó en gran medida a la señalización prolongada de NF-κB en ratones deficientes en YAP/TAZ en las células ATII, ya que se descubrió que IκBα (inhibidor α de NFκB) es un objetivo directo de YAP/TAZ y de su factor de transcripción asociado, TEAD (familia del dominio TEA). Además, la ablación de ABL1 (cinasa de Abelson-1) en células secretoras que expresan SCGB1A1 promueve la expansión de una pequeña población doblemente positiva para SCGB1A1/SPC (proteína surfactante C) para reponer las células ATI distales dañadas durante la neumonía bacteriana. Esta rápida y mejorada proliferación limitó el edema alveolar y mejoró la recuperación tras la infección por S. aureus y S. pneumoniae, sin afectar la carga bacteriana ni la respuesta de citocinas inflamatorias. Dados estos resultados, se debería prestar mayor atención a las vías de reparación epitelial como mecanismo de protección contra la neumonía grave, ya que estas vías son ampliamente aplicables a diversas etiologías.
Células endoteliales
En la interfaz alvéolo-capilar, el intercambio de gases se lleva a cabo no solo por las células ATI, como se describió anteriormente, sino también por las células endoteliales microvasculares pulmonares, que proporcionan glóbulos rojos listos para transportar oxígeno. Estas células también cumplen varias funciones importantes en respuesta a la infección respiratoria. En primer lugar, son un conducto principal para la entrada de leucocitos desde el torrente sanguíneo, proporcionando las moléculas de anclaje involucradas en la transmigración de leucocitos. En respuesta a mediadores inflamatorios, las células endoteliales también liberan el quimioatrayente de neutrófilos IL-8 en gránulos preformados, llamados cuerpos de Weibel-Palade. Si bien el papel de las células endoteliales en la transmigración de leucocitos se ha revisado ampliamente en otras publicaciones, incluyendo mecanismos específicos del pulmón, se sabe mucho menos sobre otras respuestas endoteliales directas e indirectas a la infección. En reposo, las células endoteliales proporcionan una barrera no trombogénica para prevenir la coagulación inapropiada; sin embargo, en respuesta a ligandos inflamatorios, activan las plaquetas y producen componentes de la coagulación. La activación de las células endoteliales durante la neumonía también produce vasoconstricción y un aumento de la permeabilidad, lo que conduce a un edema alveolar excesivo. Las infecciones, incluidas las causadas por el SARS-CoV-2, generan concentraciones considerables de citocinas proinflamatorias, lo que puede provocar complicaciones potencialmente mortales, como el síndrome de liberación de citocinas (SLC). Recientemente se demostró que la señalización trans-IL-6 endotelial era necesaria para la máxima expresión de IL-6, IL-8, CCL2 (ligando 2 del motivo C-C) y PAI-1 (inhibidor del activador del plasminógeno-1), todos los cuales se encuentran elevados en pacientes con SLC como resultado de sepsis, SDRA, quemaduras e infección por SARS-CoV-2. En contraste, la expresión del receptor 1 de esfingosina-1-fosfato (S1P1) en las células endoteliales puede servir para limitar la liberación excesiva de citocinas, cuyo agonismo protegió a los ratones de la infección letal por influenza a través de una reducción en la expresión de citocinas específicas del endotelio y el reclutamiento de leucocitos, sin impacto en la carga viral. La lesión endotelial que ocurre durante la infección pulmonar resulta en el reclutamiento y activación de plaquetas a través de interacciones endotelio-plaqueta. Se demostró que restringir la agregación y activación plaquetaria prevenía la hemorragia pulmonar y la trombosis en ratones infectados con influenza de una manera que dependía de una reducción en el reclutamiento de leucocitos y los niveles de citocinas a los 6 días posteriores a la infección. En este contexto, la activación plaquetaria dependía de PAR4 (receptor 4 activado por proteasa), que juega un papel en la activación plaquetaria mediada por trombina. También se ha demostrado que la activación directa de las plaquetas ocurre durante la infección por influenza a través de su fagocitosis de viriones de una manera dependiente de TLR7. La captación viral estimuló a las plaquetas a producir el componente C3 del complemento, lo que promovió la NETosis de los neutrófilos y la producción de mieloperoxidasa, que luego fue atenuada por la producción de GM-CSF por las mismas plaquetas activadas, probablemente como un mecanismo para limitar la inflamación y la trombosis.
Es importante destacar que las células endoteliales pulmonares no constituyen una población homogénea, y su heterogeneidad se ha explorado recientemente mediante la secuenciación de ARN de células individuales. Se identificaron cinco grupos de células endoteliales, que representan células endoteliales macrovasculares, tres grupos de células endoteliales microvasculares y un nuevo subconjunto endotelial con alta expresión de Car4 que se yuxtapone a las células ATI tras una lesión pulmonar aguda (inducida por influenza o bleomicina). Si bien no es específico de las células endoteliales con alta expresión de Car4, se predijo que se producirían interacciones endotelio-epiteliales entre las células de tipo I y múltiples subpoblaciones de células endoteliales altamente proliferativas tras la infección por influenza. Al igual que las células epiteliales, los mecanismos de reparación endotelial se reconocen como un proceso igualmente importante para limitar la lesión pulmonar inducida por neumonía. COUP-TF2 (factor de transcripción 2 del promotor corriente arriba de la ovoalbúmina de pollo) se ha identificado como un factor de transcripción crucial para la proliferación de células endoteliales mediante la activación de la proteína del ciclo celular ciclina D1 (CCND1) y la promoción de la angiogénesis a través de la activación de neuropilina-1 mediada por VEGFA/VEGFR2 (factor de crecimiento endotelial vascular A/receptor 2 del factor de crecimiento endotelial vascular). La eliminación de COUP-TF2 redujo la capacidad proliferativa de las células endoteliales ex vivo e in vivo, lo que resultó en una lesión pulmonar exacerbada y una mayor mortalidad en ratones infectados con influenza. Curiosamente, este estudio corrobora el hallazgo previo de que las células endoteliales supervivientes ubicadas cerca de áreas de lesión epitelial exhiben la mayor capacidad de proliferación. Estos estudios resaltan la importancia de las células estructurales, como las células endoteliales, epiteliales y los fibroblastos, en la defensa del huésped y la protección contra la lesión pulmonar inducida por neumonía. Los estudios actuales sobre células endoteliales se limitan en gran medida a lo que ocurre tras una infección viral (influenza, SARS-CoV-2), y los estudios futuros deberían ampliarse a la infección por patógenos bacterianos o fúngicos, lo que podría demostrar la amplia relevancia de los mecanismos específicos del endotelio durante la infección pulmonar.
Fibroblastos y matriz extracelular
La función principal de la matriz extracelular (MEC) pulmonar es anclar y proporcionar soporte estructural al epitelio y endotelio pulmonares. Los componentes de la MEC son sintetizados y mantenidos en gran parte por fibroblastos, con contribuciones menores de células endoteliales y epiteliales. En términos generales, la MEC se puede subdividir en matriz intersticial y membrana basal, que en conjunto comprenden 150 proteínas estructurales esenciales en el pulmón. La membrana basal está compuesta por glicoproteínas en una fina capa en la superficie basal de las células, mientras que la matriz intersticial es una red laxa de colágenos fibrilares (I, II, III, V, XI), fibras elásticas (elastina reticulada con una capa externa de microfibrillas), fibrilinas, glicoproteínas, proteoglicanos (PG) y glicosaminoglicanos (GAG) ubicados entre las membranas basales.
En el pulmón, el glicosaminoglicano hialuronano (HA) y los proteoglicanos (PG) unidos a él constituyen componentes estructurales de la matriz intersticial e importantes reguladores de las respuestas inmunitarias pulmonares. En condiciones basales, el HA se sintetiza principalmente como HA de alto peso molecular (HA-PM), cuya función es mantener la homeostasis pulmonar y limitar las respuestas inflamatorias. Los PG están compuestos por un núcleo proteico central (en el pulmón, con mayor frecuencia versicano, biglicano y decorina), entrecruzado con cadenas laterales de GAG con carga negativa (como el sulfato de condroitina o el sulfato de heparina) o HA-PM. Esta compleja estructura matricial, junto con la carga negativa de las cadenas de GAG, forma un gel hidratado que rodea a los demás componentes estructurales de la matriz extracelular (colágeno y elastina), así como a múltiples proteínas solubles, incluyendo quimiocinas, citocinas, factores de crecimiento y proteínas modificadoras de la matriz. La complejidad de esta estructura permite el secuestro de estas diversas proteínas, posibilitando un control rápido y preciso de las respuestas inmunitarias.
Durante una lesión o inflamación, el HA se escinde en HA de bajo peso molecular (HA-BM) mediante la acción directa de hialuronidasas, ROS o daño tisular. El HA-BM es altamente proinflamatorio y puede inducir la producción de citocinas y quimiocinas proinflamatorias, lo que conduce a un mayor reclutamiento celular. Además, las quimiocinas de neutrófilos se unen a los GAG pulmonares con afinidad variable, generando gradientes de quimiocinas funcionalmente diferentes para coordinar las velocidades de migración de los neutrófilos a través de la ECM. La producción del PG versicano aumenta en la inflamación aguda y crónica, lo que conduce al secuestro y estabilización de citocinas y quimiocinas, activación de la señalización TLR y modificación de la estructura general de la ECM para afectar la migración celular. En un modelo de influenza de neumonía, los fibroblastos que responden al daño produjeron la proteasa ADAMTS4 (una desintegrina y metaloproteinasa con motivos de trombospondina 4), lo que condujo a la escisión del versicano, permitiendo la infiltración de células inmunes. En general, la matriz extracelular (MEC) desempeña un papel fundamental en las respuestas inmunitarias innatas, pero nuestra comprensión de la compleja interacción de los componentes de la MEC y la contribución de las células circundantes está aún en sus inicios.
Conclusión
La respuesta inmune innata en el pulmón es un componente complejo y sofisticado de respuesta rápida del sistema inmunitario. Dada la naturaleza rápida y temprana de las respuestas inmunes innatas, es probable que el futuro del descubrimiento de terapias para la neumonía implique una modulación precisa y personalizada de estas respuestas para maximizar la eliminación de patógenos y minimizar el daño tisular secundario. Si bien muchos aspectos del sistema inmunitario innato se conocen desde hace tiempo, dos avances recientes en este campo han transformado radicalmente nuestra comprensión de este sistema en el pulmón. El surgimiento de tecnologías basadas en células individuales ha revelado variaciones y heterogeneidad en las respuestas inmunes innatas, específicas de cada órgano y tipo celular. Estas nuevas bases de datos ómicas de células individuales proporcionan enormes cantidades de datos específicos del pulmón relacionados con la inmunidad innata. El segundo avance importante se relaciona con la reciente pandemia de SARS-CoV-2, donde la recolección de muestras y datos clínicos permitió un nivel de detalle sin precedentes con respecto a las respuestas inmunes innatas y adaptativas a un único patógeno pulmonar. Hasta el momento, no se ha percibido el impacto total de estas nuevas fuentes de información en nuestra comprensión de la inmunidad innata. Esperamos que estos dos conjuntos de datos sirvan de base para innumerables descubrimientos sobre la biología de las respuestas inmunitarias innatas pulmonares y conduzcan al desarrollo de nuevas terapias inmunomoduladoras para el tratamiento de la neumonía.

