Historia del lactato
La acidosis láctica en el contexto de enfermedades graves tiene una historia que se remonta al siglo XIX, cuando Johann Joseph Scherer midió por primera vez los niveles de ácido láctico en sangre post mortem de dos mujeres que murieron de fiebre puerperal. Posteriormente, Folwarczny en 1858 describió niveles elevados de lactato en un paciente vivo con leucemia, y más tarde Salomon en 1878 observó un aumento de los niveles de lactato en pacientes con enfermedad pulmonar obstructiva crónica, neumonía, tumores sólidos e insuficiencia cardíaca congestiva. Varios años después, Fletcher describió cómo el ácido láctico era producido por el músculo esquelético en condiciones anaeróbicas, señalando que cuando el oxígeno estaba fácilmente disponible, «o restringe mediante alguna guía del evento químico la producción de ácido en el músculo, o es capaz de eliminarlo después de su producción». Estas observaciones realizadas hace más de 100 años representan la base sentada para la comprensión del ácido láctico en los estados de enfermedad de los pacientes críticamente enfermos.
A finales de la década de 1950, Huckabee realizó una serie de importantes experimentos fisiológicos, resumiendo la relación de los niveles de lactato y piruvato en sangre con diversos estados de deficiencia de oxígeno, incluyendo el ejercicio extremo, la respiración de gases con baja tensión de oxígeno y la disminución del gasto cardíaco. Continuó demostrando niveles elevados en pacientes en diversas etapas de shock. Casi dos décadas después, Woods y Cohen crearon un esquema de clasificación de la acidosis láctica basado en el trabajo original de Huckabee, designando el tipo A como aquel que surge de la disminución de la perfusión u oxigenación y el tipo B derivado de enfermedades subyacentes, medicamentos/intoxicaciones o errores innatos del metabolismo. Este esquema continúa utilizándose en la actualidad como un medio para clasificar y comprender los orígenes del lactato elevado. Estudios posteriores sobre el metabolismo del lactato ampliaron las visiones sobre el uso del lactato en el cuerpo. En la década de 1980, se postuló por primera vez la idea de las lanzaderas de lactato y del lactato mismo como fuente de energía. El lactato ya no se consideraba un subproducto final sin salida del metabolismo, sino una fuente normal y, en ocasiones, preferida de combustible metabólico.
Metabolismo del lactato
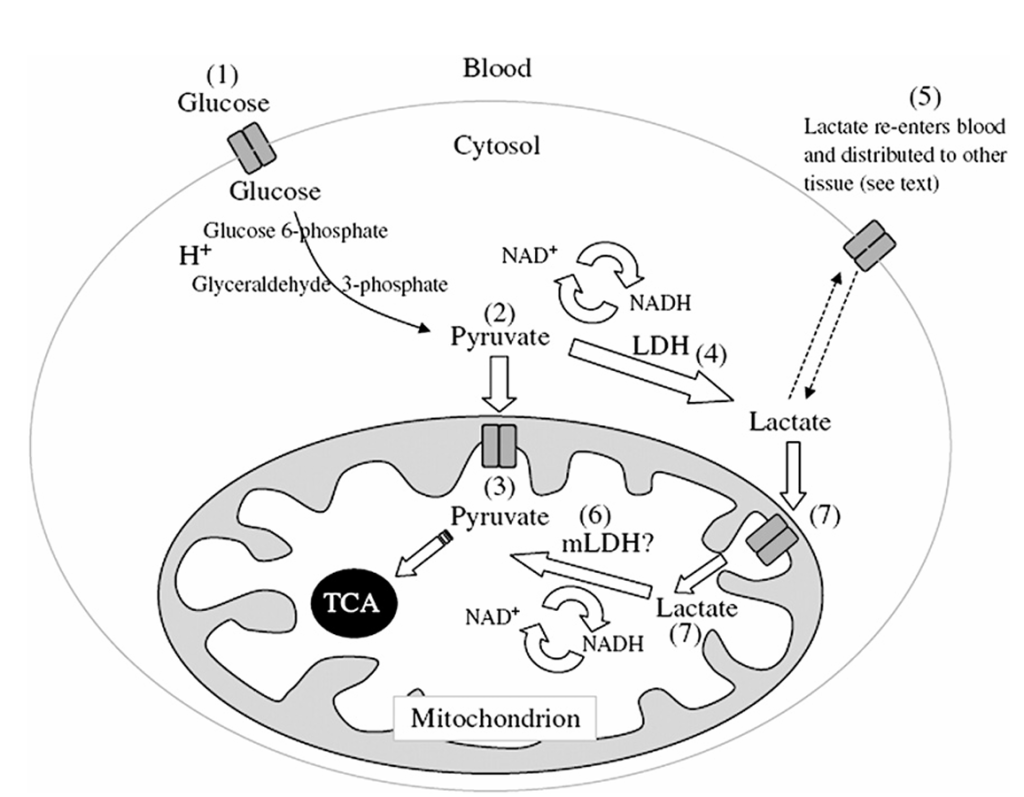
El lactato se forma a partir de la reducción del piruvato mediante la enzima lactato deshidrogenasa:Este proceso produce dos moléculas de ATP, lo que convierte la formación de lactato en una fuente de energía celular durante el metabolismo anaeróbico. La reacción ocurre dentro del citosol como el paso final de la glucólisis. En un estado fisiológico basal, la reacción favorece la formación de lactato a partir de piruvato en una proporción aproximada de 10:1. La reducción del piruvato es la única vía conocida para la producción de lactato, lo que convierte esto en una forma única de monitorear los procesos metabólicos anaeróbicos (Fig. 1).
Los niveles de lactato en sangre resultan del equilibrio entre la producción y la eliminación, una fuente de importante interés científico en las últimas décadas. Normalmente, los niveles de lactato en sangre son inferiores a 2mmol/L. En condiciones fisiológicas normales, se producen aproximadamente 1500 mmol de lactato al día, principalmente del músculo esquelético, la piel, el cerebro, el intestino y los glóbulos rojos. En enfermedades graves, la producción de lactato ocurre en muchos otros tejidos. Los pulmones, por ejemplo, pueden ser una fuente significativa de lactato durante la lesión pulmonar aguda a pesar de la ausencia de hipoxia tisular. Los leucocitos también pueden producir grandes cantidades de lactato durante la fagocitosis o cuando se activan en la sepsis. Los órganos esplácnicos, como el hígado y los intestinos, son otra fuente potencial de producción de lactato y pueden ser particularmente vulnerables a una vasoconstricción desproporcionada en estados de bajo flujo. Si este mecanismo contribuye o no al aumento del lactato intestinal en la sepsis sigue siendo un tema de debate. De Backer y colegas mostraron, mediante oximetría venosa hepática, que solo 6 de 90 pacientes con sepsis grave tenían producción esplácnica de lactato, incluso en aquellos con niveles séricos de lactato gravemente elevados.
La eliminación del lactato ocurre principalmente en el hígado con contribuciones importantes del riñón %) en menor medida de otros órganos (corazón y músculo esquelético). La utilización ocurre a través del ciclo de Cori, donde el lactato se convierte de nuevo en piruvato y finalmente en glucosa mediante la gluconeogénesis. Se ha demostrado que en pacientes con enfermedad hepática crónica (generalmente encefalopatía grado III o IV), la eliminación del lactato está disminuida, contribuyendo así también a niveles sanguíneos elevados. Además de los mecanismos de eliminación metabólica, el lactato puede ser excretado por el riñón una vez que se supera el umbral renal (aproximadamente ). Por lo tanto, la insuficiencia hepática y renal puede alterar la eliminación del lactato.
La acidosis láctica está típicamente presente en estados de shock en los que el suministro de oxígeno tisular (DO2) es insuficiente para satisfacer la demanda celular. En esta acidosis láctica tipo A clásica, el flujo a través de la vía glucolítica aumenta, lo que lleva a una acumulación de piruvato. En un estado de baja tensión de oxígeno, el piruvato no entra en la mitocondria para la fosforilación oxidativa. Se sabe que la hipoxia inhibe el complejo de la piruvato deshidrogenasa (PDH) involucrado en la descomposición aeróbica del piruvato en acetil coenzima A (CoA) para su entrada en el ciclo de Krebs. También se sabe que inhibe la piruvato carboxilasa, que convierte el piruvato en oxaloacetato al principio del proceso de gluconeogénesis. Esto provoca una rápida acumulación de piruvato, y el metabolismo del piruvato se desplaza posteriormente casi en su totalidad hacia la formación de lactato. En consecuencia, la concentración intracelular de lactato aumenta rápidamente, lo que lleva a su excreción al torrente sanguíneo. La formación clínicamente significativa de lactato por bajo flujo se demostró de manera más notable en un grupo de pacientes con shock cardiogénico por Levy y colegas donde las proporciones lactato:piruvato se calcularon en 40:1 en comparación con los controles (10:1). Más evidencia del aumento de la producción de lactato durante estados de shock provino de Revelly y colegas, quienes compararon siete pacientes con shock cardiogénico y siete pacientes con shock séptico con siete controles sanos. Al infundir lactato marcado con C y glucosa marcada con H de forma continua, mostraron que la hiperlactatemia resultaba de una sobreproducción de lactato y que la eliminación era similar en los tres grupos.
La producción excesiva de lactato puede no ser el único contribuyente a la hiperlactatemia en pacientes críticamente enfermos. Levraut y colegas mostraron que en pacientes con sepsis hemodinámicamente estable, los niveles elevados de lactato están más relacionados con una eliminación alterada que con una sobreproducción. El metabolismo general del lactato en enfermedades críticas es, por lo tanto, un proceso altamente complejo con muchos factores que influyen en los niveles de lactato en sangre. El lactato en sí mismo probablemente no es dañino y es transportado a los tejidos durante estados de estrés como combustible energético de esqueleto carbonado. Cuando los niveles de lactato están elevados en la sangre, puede ser más un indicador de un estado de estrés subyacente y no necesariamente la causa directa de la patogénesis.
Papel como marcador pronóstico en pacientes críticamente enfermos
Ya sea que los niveles de lactato en sangre estén elevados debido al aumento de la producción o a la disminución de la eliminación, el monitoreo de los niveles puede resultar valioso como biomarcador de un estado subyacente de enfermedad crítica, como el shock. El lactato es uno de los muchos marcadores utilizados para el pronóstico en pacientes críticamente enfermos y la campaña de supervivencia a la sepsis recomienda un valor superior a 4mmol/L como sugestivo de sepsis grave que requiere reanimación agresiva.
Huckabee realizó los primeros análisis de niveles elevados de lactato en pacientes con diversos grados de shock. Reportó una serie de casos de nueve pacientes con acidosis láctica que se quejaban de hiperpnea y disnea. El síndrome clínico progresó a debilidad, estupor y muerte durante varias etapas de otras enfermedades graves (es decir, postgastrectomía, poliomielitis, neumonía y endocarditis bacteriana). Encontró un amplio rango de niveles de lactato (3-26 mmol/L) en varios días de evolución de su enfermedad. Observó que los niveles elevados de lactato indicaban hipoxia tisular generalizada pero sin una causa aparente de la hipoxia. En su artículo, afirma: «el síndrome químico pudo reproducirse en animales solo mediante insuficiencia circulatoria periférica gradual y este síndrome no pudo descartarse en los pacientes».
Para la década de 1970, Weil y Afifi y Cady y colegas ampliaron los experimentos de Huckabee mostrando prospectivamente que el lactato era un fuerte predictor de muerte en pacientes críticamente enfermos. La muerte ocurrió en dos tercios de los pacientes con niveles de lactato superiores a 3.8mmol/L y se acercó al a medida que los niveles se acercaban a 8mmol/L. Más tarde, en 1994, Stacpoole y colegas informaron sobre la historia natural del lactato elevado en pacientes médicos y quirúrgicos críticamente enfermos, demostrando que los niveles elevados de lactato (>5mmol/L) predecían la muerte a lo largo del tiempo. La supervivencia fue del , y 17% a 1, 3 y 30 días, respectivamente, para pacientes con niveles de lactato persistentemente anormales. La supervivencia media global en este grupo fue de 2 días.
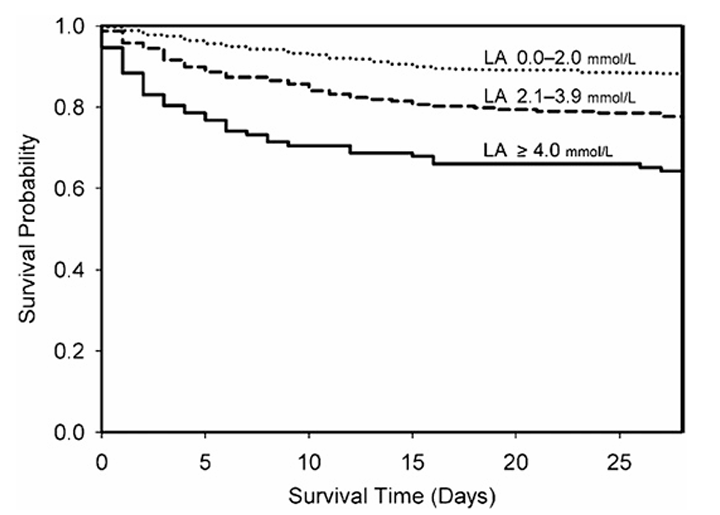
Más recientemente, en 2007, Trzeciak y colegas observaron los niveles iniciales de lactato sérico en más de 1100 pacientes vistos en un departamento de emergencias (ED), una unidad de cuidados intensivos (ICU) y salas de hospital general. Los niveles de lactato se dividieron en bajo (0-2 mmol/L), intermedio (2.1-3.9 mmol/L) y alto (). Encontraron que un nivel de lactato superior a 4mmol/L era altamente específico para predecir la fase aguda de la muerte y la muerte intrahospitalaria en los tres grupos (Fig. 2). Concluyeron que los niveles iniciales de lactato (ante la sospecha de sepsis clínica) podrían usarse para aumentar, pero no reemplazar, la evaluación de la mortalidad al lado de la cama, independientemente de la ubicación del paciente (ED, sala o ICU).
Shapiro y colegas tomaron pacientes con sospecha de infección en un ED e hipotetizaron que los niveles iniciales de lactato sérico predecirían la mortalidad hospitalaria. Nuevamente, los niveles de lactato se estratificaron en bajo , intermedio y alto . Mostraron una probabilidad creciente de mortalidad y , respectivamente) para cada grupo y calcularon una especificidad del para la muerte. Más tarde, en 2007, el grupo de Shapiro28 incluyó pacientes normotensos e hipotensos que se presentaban en un ED con sospecha de infección. Mostraron una odds ratio de muerte de 2.2 para aquellos pacientes con niveles de lactato intermedios y una odds ratio de 7.1 para niveles altos de lactato. Estos valores eran independientes de la hipotensión (Fig. 3). La mortalidad en los pacientes normotensos con niveles de lactato superiores a fue similar a la de los pacientes normotensos que tenían presiones arteriales sistólicas inferiores a .
(De Springer Science1Business Media. Howell MD, Donnino M, Clardy P, et al. Occult
hypoperfusion and mortality in patients with suspected infection.
La pregunta sigue siendo: ¿Es el nivel inicial de lactato un verdadero biomarcador de estratificación de riesgo o solo una manifestación de disfunción orgánica? Mikkelsen y colegas realizaron una reciente cohorte unicéntrica de 830 pacientes con sepsis grave ingresados a través de un ED. Observaron a pacientes con shock y sin shock con niveles bajos , intermedios y altos (>4mmol/L) de lactato. Encontraron que el nivel inicial de lactato sérico predecía la mortalidad en ambos grupos y hallaron que la mortalidad era del 15.4%, 37% y 46.9% en los grupos de lactato bajo, medio y alto con shock séptico, respectivamente. También encontraron una mortalidad del 8.7%, 16.4% y 31.8% en los grupos sin shock, respectivamente. Estos valores se calcularon después de la corrección por disfunción orgánica con las puntuaciones APACHE II, mostrando el poder predictivo del nivel inicial de lactato de forma única como biomarcador. Se observó una mortalidad relativamente alta en pacientes sépticos sin shock con niveles de lactato relativamente intermedios (2.5−4mmol/L). Shapiro y Mikkelsen demostraron mortalidades predichas en estos pacientes de aproximadamente 15%. Los pacientes normotensos con presunta sepsis y niveles de lactato intermedios-altos probablemente representan un subgrupo de alto riesgo en quienes la reanimación temprana y agresiva puede mejorar la mortalidad. La campaña de supervivencia a la sepsis recomienda la terapia temprana dirigida por objetivos en individuos con sepsis grave o shock séptico, particularmente si el nivel de lactato es superior a 4mmol/L. Jansen y colegas mostraron que los niveles de lactato en sangre estaban fuertemente asociados con las puntuaciones SOFA, especialmente al principio de la estancia en la ICU. Encontraron que los niveles iniciales de lactato entre 2 y 3mmol/L correspondían a una mortalidad del 60%. Quizás el inicio de la terapia temprana dirigida por objetivos, según lo recomendado por la campaña de supervivencia a la sepsis, debería ampliarse para incluir a aquellos pacientes con presunta sepsis y niveles de lactato intermedios (2−4mmol/L). Los niveles de lactato sérico como biomarcador predictivo pueden resultar más útiles en esta población.
Relaciones lactato: piruvato
Las relaciones lactato:piruvato se han empleado en el pronóstico y para distinguir la hiperlactatemia tipo A de la tipo B. Sin embargo, los ensayos de piruvato no siempre están disponibles y pueden ser inexactos si la muestra se hemoliza, se almacena incorrectamente o no se procesa dentro de las primeras 3 horas. Un ejemplo de su utilidad se puede encontrar en un estudio de Levy y colegas en el que estaban presentes niveles elevados de lactato en asociación con relaciones lactato:piruvato aumentadas en los no supervivientes (37:1) en relación con los supervivientes (20:1) en pacientes con shock séptico. Suistomaa y colegas también midieron los niveles de lactato y las relaciones lactato:piruvato durante las primeras 24 horas en pacientes médico-quirúrgicos críticamente enfermos. El nivel inicial elevado de lactato, la elevación continua del lactato y la relación lactato:piruvato predijeron la mortalidad. En pacientes con sepsis grave, la elevación del lactato se asoció con relaciones lactato:piruvato normales, mientras que en la insuficiencia circulatoria, ambos estaban elevados. Esto apoya en cierta medida las teorías de que las relaciones lactato:piruvato elevadas apoyan un mecanismo hipóxico de producción de lactato. Las relaciones normales de lactato a piruvato en el contexto de niveles elevados de lactato pueden significar un mecanismo no hipóxico. Se necesitan más estudios. Con las muchas dificultades para obtener niveles precisos de piruvato, es dudoso que medir la relación lactato:piruvato añada algún valor pronóstico adicional para la reanimación del shock.
Depuración de lactato
Las mediciones únicas de lactato sérico pueden tener limitaciones y quizás las mediciones en serie mejoren la capacidad pronóstica. Se demostró que la depuración del lactato a lo largo del tiempo era superior a las variables derivadas del oxígeno y consumo de oxígeno en pacientes con shock séptico. Los niveles de lactato en fase inicial y final fueron más bajos en los supervivientes, mientras que y no fueron diferentes. Bakker y colegas mostraron que el lactime, o la duración de los niveles elevados de lactato sérico, se correspondía con mayor precisión con la insuficiencia orgánica y la muerte, y lo hacía mejor que los niveles iniciales de lactato, y . Abramson y colegas demostraron que todos los pacientes traumatizados que normalizaron sus niveles de lactato sérico en 24 horas sobrevivieron y que aquellos que los depuraron en 48 horas tenían una probabilidad de supervivencia del 75%. La capacidad de normalizar el lactato a un valor inferior a 2 mmol/L predijo la supervivencia (P<.0001), mientras que y no.
Estos hallazgos fueron corroborados por McNelis y colegas, quienes demostraron una mortalidad del 100% en pacientes de una ICU quirúrgica que tenían niveles de lactato persistentemente elevados. Aquellos que depuraron su lactato (nivel de lactato <2 mmol/L) en las primeras 24 horas tuvieron una mortalidad del 3.9%. Los pacientes que tuvieron una depuración retardada del lactato (>48 horas para alcanzar un nivel de lactato <2 mmol/L) tuvieron una mortalidad del 42.5%. Husain y colegas reforzaron la importancia de la depuración del lactato en pacientes críticamente enfermos de una ICU quirúrgica cuando estratificaron el riesgo de 95 pacientes traumatizados y no traumatizados en cuatro grupos según su capacidad para depurar el lactato: (1) depuración en las primeras 24 horas, (2) depuración en 24 a 48 horas, (3) más de 48 horas para normalizar o (4) nunca normalizado. La mortalidad predicha se calculó como 10%, 20%, 23% y 67%, respectivamente, en los cuatro grupos. Las mediciones iniciales y seriadas de lactato predijeron la supervivencia con significación estadística.
En una población de pacientes diferente, Nguyen y colegas38 cuantificaron la depuración del lactato en 111 pacientes con sepsis grave y shock séptico. Los supervivientes tuvieron una depuración de lactato del 38% frente al 12% de los no supervivientes. Una baja depuración de lactato (<10%) dentro de las primeras 6 horas predijo la muerte en dos tercios de los casos. No está claro que ninguna intervención ayude a mejorar la depuración del lactato, pero quizás las mediciones seriadas de lactato podrían usarse como marcadores de progreso en la reanimación del shock.
Durante muchos años, se pensó que el lactato en sí mismo era dañino y contribuía al empeoramiento de la acidosis. Desde entonces se ha demostrado que esto probablemente no es cierto. Sin embargo, en un esfuerzo por reducir activamente los niveles de lactato, Stacpoole y colegas realizaron una serie de experimentos con dicloroacetato (DCA). El DCA estimula el complejo PDH uniéndose e inhibiendo la quinasa de PDH, que inactiva la enzima PDH. Aumentar el flujo a través de la vía enzimática de la PDH parecía una forma ideal de reducir los niveles de lactato y se ha estudiado en una variedad de poblaciones de pacientes: niños con acidosis láctica congénita, pacientes con isquemia miocárdica, y pacientes críticamente enfermos con shock. Todos los estudios han demostrado que el DCA reduce de forma segura los niveles circulantes de lactato en sangre. Sin embargo, el único ensayo controlado de DCA, realizado por Stacpoole y colegas, para el tratamiento de la acidosis láctica mostró una disminución de los niveles de lactato pero ningún cambio en ninguna medición hemodinámica significativa o en la supervivencia. Muchos pacientes fueron incluidos con niveles de lactato significativamente elevados y muchos fueron incluidos tarde en sus estados de shock, ya en desarrollo de insuficiencia orgánica múltiple. Sin embargo, el DCA nunca ha demostrado ser útil en el tratamiento de pacientes críticamente enfermos con niveles elevados de lactato.
Lactato arterial versus venoso
Para comparar el lactato arterial y venoso, se extrajeron muestras de lactato arterial y venoso a 74 pacientes adultos de un ED con una diferencia de 5 minutos entre ellas.44 La correlación entre el lactato arterial y venoso fue de 0.94 (IC 95%, 0.91-0.96). Hubo una diferencia media de lactato venoso menos arterial de 0.22 mmol/L (IC 95%, 0.04-0.41), que varió de -1.3 a 1.7 mmol/L en pacientes individuales. De los pacientes de la muestra, 30% tenían niveles de lactato arterial inferiores a 1.6.
En un estudio de pacientes traumatizados, se tomaron mediciones de lactato arterial y venoso, extraídas con una diferencia de 2 minutos entre sí en 221 pacientes. Los niveles se correlacionaron con una correlación de 0.94 (IC 95%, 0.94-0.96; P=.0001). La ecuación para la diferencia en los valores se expresó como lactato arterial =0.0706+0.889 (lactato venoso). La diferencia entre los valores arteriales y venosos no fue estadísticamente significativa.
Solución de ringer lactato
Algunos proveedores han expresado su preocupación sobre el uso de la solución de Ringer lactato en el contexto de la acidosis láctica, teorizando que podría empeorar la acidosis láctica. Un litro de solución de Ringer lactato contiene 130 mEq de sodio, 4mEq de potasio, 3 mEq de calcio, 109 mEq de cloruro y 28 mEq de lactato mezclados en agua estéril. El contenido de electrolitos es isotónico (273 mOsm/L, calculado) en relación con el líquido extracelular (aproximadamente 280 mOsm/L). El agua estéril es ácida (pH 5 a 7) como resultado de las interacciones con el aire y la bolsa de plástico. La adición de lactato de sodio aumenta el pH a aproximadamente 6.6 (rango 6.0-7.5). Por lo tanto, el lactato actúa como una base y como tal no puede causar acidosis. A menudo, debido al diagnóstico de acidosis láctica, los médicos eligen solución salina para los líquidos de reanimación. En un estudio de sesenta pacientes con sepsis grave o shock séptico, la acidosis metabólica hiperclorémica debida a la infusión de solución salina fue la causa predominante de acidosis metabólica. Además, para determinar si la solución de Ringer lactato aumentaba las concentraciones circulantes de lactato, se administraron infusiones de 1 L de solución de Ringer lactato o dextrosa al 5% durante 1 hora a voluntarios adultos sanos. Las concentraciones de lactato no fueron significativamente diferentes entre los dos grupos. Por lo tanto, los niveles elevados de lactato en sangre en el contexto de la infusión de solución de Ringer lactato son un hallazgo inesperado y no deben atribuirse a la infusión.
En resumen, la comprensión de la acidosis láctica tipo A, el lactato sérico inicial, las mediciones seriadas de lactato y la depuración del lactato pueden ser útiles en el manejo de pacientes críticamente enfermos. Los niveles elevados de lactato probablemente estén relacionados con un aumento de la producción y una disminución de la eliminación, dependiendo de los complejos factores metabólicos de cada paciente. Los niveles de lactato venoso son fáciles de obtener, económicos y pueden proporcionar información valiosa en el pronóstico de pacientes médicos y quirúrgicos con shock.
Acidosis láctica tipo B
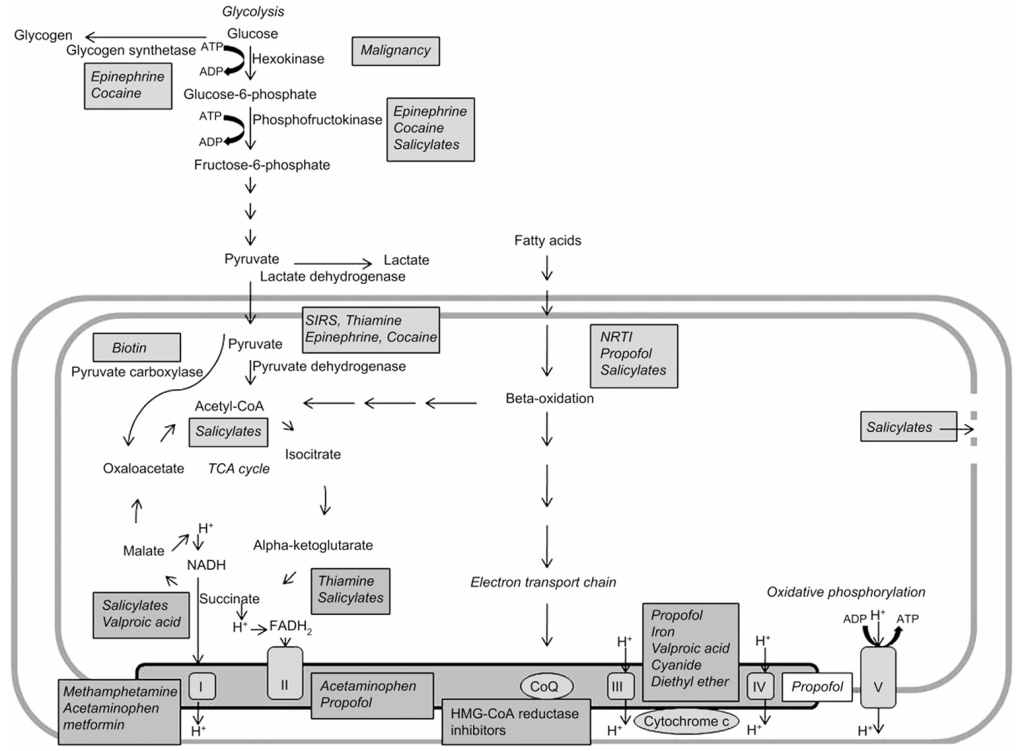
A menudo, durante el curso de una enfermedad crítica, los pacientes tienen elevaciones continuas del nivel de lactato sin evidencia continua de hipoxia o isquemia celular (Fig. 4). En estos casos es importante considerar lo que Woods y Cohen denominaron acidosis láctica tipo B. El Recuadro 1 enumera las causas de la acidosis láctica tipo B. La acidosis láctica tipo B se divide en tipo B1 (relacionada con enfermedades subyacentes), tipo B2 (relacionada con el efecto de fármacos y toxinas) y tipo B3 (asociada con errores innatos del metabolismo).
Recuadro 1. Causas de la acidosis láctica tipo B
Tipo B1 — Enfermedades subyacentes
- Insuficiencia renal
- Insuficiencia hepática
- Diabetes mellitus
- Neoplasias malignas
- Síndrome de respuesta inflamatoria sistémica (SRIS)
- Virus de la inmunodeficiencia humana (VIH)
Tipo B2 — Fármacos y toxinas
- Acetaminofén (paracetamol)
- Alcoholes — etanol, metanol, dietilenglicol, isopropanol y propilenglicol
- Análogos nucleósidos antirretrovirales — zidovudina, didanosina y lamivudina
- Agonistas β-adrenérgicos — epinefrina, ritodrina y terbutalina
- Biguanidas — fenformina y metformina
- Cocaína, metanfetamina
- Compuestos cianogénicos — cianuro, nitrilos alifáticos y nitroprusiato
- Éter dietílico
- Fluorouracilo
- Halotano
- Hierro
- Isoniazida
- Linezolid
- Ácido nalidíxico
- Niacina
- Propofol
- Salicilatos
- Estricnina
- Azúcares y polioles — fructosa, sorbitol y xilitol
- Sulfasalazina
- Nutrición parenteral total
- Ácido valproico
- Deficiencias vitamínicas — tiamina y biotina
Tipo B3 — Errores congénitos del metabolismo
- Deficiencia de glucosa-6-fosfatasa (enfermedad de Von Gierke)
- Deficiencia de fructosa-1,6-difosfatasa
- Deficiencia de piruvato carboxilasa
- Deficiencia del complejo piruvato deshidrogenasa (PDH)
- Aciduria metilmalónica
- Síndrome de Kearns-Sayre
- Síndrome de Pearson
- Encefalomiopatía mitocondrial con acidosis láctica y episodios tipo ictus (MELAS)
- Epilepsia mioclónica con fibras rojas rasgadas (MERRF)
Enfermedades subyacentes de tipo B
Acidosis Láctica Asociada a Malignidad
En la década de 1920, Warburg midió el VO2 y la producción de lactato en células tumorales en condiciones aeróbicas y anaeróbicas. Encontró que las células tumorales tenían un alto consumo de glucosa y producción de lactato en presencia de oxígeno. Creía que esta «glucólisis aeróbica» se debía a una función anormal de las mitocondrias y era la raíz de la transformación maligna. Un factor que contribuye a la alta tasa de glucólisis es la sobreexpresión de enzimas glucolíticas, como la hexoquinasa. En contraste con la hipótesis de Warburg, la literatura respalda que, excepto en algunos cánceres, la alteración mitocondrial en los tumores es el resultado de cambios metabólicos relacionados con el tumor y no la etiología de la malignidad. Además, no todos los tumores utilizan la glucólisis como la forma preferida de producción de energía. Sin embargo, la acidosis láctica se ha informado con mayor frecuencia en neoplasias hematológicas, como leucemias y linfomas, y también se ha informado en melanoma, cáncer de pulmón de células pequeñas, mieloma múltiple, sarcoma, cáncer de mama, carcinoma de células oat, carcinoma indiferenciado, colangiocarcinoma y feocromocitoma (aunque esto probablemente se deba a las catecolaminas circulantes altas [discutido más adelante]). En una revisión de la acidosis láctica en neoplasias hematológicas, los niveles de lactato informados oscilaron entre 5 y 38 mmol/L con una mortalidad informada del 93% al 96%. El manejo de la hiperlactatemia depende del tratamiento de la malignidad subyacente. Los enfoques para el manejo agudo de la acidosis láctica en estos casos incluyen el uso de insulina intravenosa para aumentar la conversión de piruvato a acetil CoA, facilitando así la oxidación de lactato a piruvato y la terapia con bicarbonato para amortiguar la acidosis extrema. Sin embargo, se ha demostrado que el bicarbonato aumenta la producción de lactato en pacientes con acidosis láctica crónica asociada a malignidad.
Síndrome de Respuesta Inflamatoria Sistémica
El síndrome de respuesta inflamatoria sistémica se asocia típicamente con acidosis láctica tipo A debido a la suposición de que la inestabilidad hemodinámica conduce a una inadecuada. Aunque esto es probablemente al menos un contribuyente parcial, hay evidencia de que el aumento de la producción de piruvato, la disminución de la actividad de la PDH (en parte debida al aumento de la quinasa de PDH), la producción de lactato por el pulmón y la disminución de la depuración de lactato son contribuyentes a la acidosis láctica en el síndrome de respuesta inflamatoria sistémica.
Insuficiencia Hepática
En pacientes críticamente enfermos con cirrosis, la acidosis láctica presagia un pronóstico sombrío con una odds ratio de 7.64 (IC 95%, 3.01-19.34) para la mortalidad en la ICU. Los individuos con la combinación de cirrosis, acidemia, acidosis láctica e insuficiencia renal aguda tuvieron una mortalidad en la ICU del 86% y una mortalidad hospitalaria del 94%. La insuficiencia hepática se asocia con una disminución de la depuración de lactato, que se exacerba aún más en la sepsis. En casos de insuficiencia hepática grave, el hígado puede ser una fuente de producción de lactato. Cuando se agregaron mediciones de lactato a los criterios del King’s College Hospital para determinar el resultado después de la intoxicación por paracetamol, un lactato temprano superior a 3.5mmol/L o un lactato posterior a la reanimación superior a 3.0mmol/L aumentaron la sensibilidad para predecir la muerte al 95%, mientras que la especificidad permaneció relativamente sin cambios en más del 90%.
Tipo B2 – fármacos y toxinas
Acetaminofén
El acetaminofén y su metabolito tóxico, la N-acetil-p-benzoquinonimina, interfieren con la fosforilación oxidativa mitocondrial al inhibir la respiración celular en sustratos vinculados a NADH (sitio de acoplamiento de energía I) y en sitios estimulados por succinato (sitio de acoplamiento de energía II). En un modelo animal, la inhibición de la respiración mitocondrial precedió a la necrosis hepática manifiesta y fue completamente prevenida por el tratamiento con N-acetil-L-cisteína. La inhibición de la fosforilación oxidativa eventualmente resulta en un cambio hacia la producción de lactato. Esto sugiere que cuanto antes se inicie el tratamiento con N-acetil-L-cisteína, mejor será el resultado.
Alcoholes Tóxicos
Las ingestiones de alcoholes tóxicos como etanol, metanol, etilenglicol y dietilenglicol pueden causar hiperosmolalidad y acidosis láctica. La intoxicación por propilenglicol es la intoxicación alcohólica más común en las UCIs. El propilenglicol es el vehículo portador de varios medicamentos intravenosos utilizados en las UCIs, que incluyen lorazepam, diazepam, digoxina, hidralazina, pentobarbital, fenobarbital, nitroglicerina, etomidato, fenitoína, multivitaminas, esmolol y trimetoprim-sulfametoxazol (Recuadro 2). El propilenglicol es oxidado por la alcohol deshidrogenasa en el hígado a lactato y piruvato. Hay muchos informes de casos de toxicidad por propilenglicol, manifestada como un hiato aniónico inexplicable, acidosis metabólica inexplicable, lactato elevado e hiperosmolalidad. La mayoría de los informes de casos han involucrado el uso de lorazepam, probablemente debido a la concentración relativamente más alta de propilenglicol en la solución. Una solución estándar de lorazepam de 2 mg/mL contiene 830 mg/mL de propilenglicol. Una dosis diaria de propilenglicol de 25 mg/kg de peso corporal se considera segura. En pacientes que requieren más de 1mg/kg/día de lorazepam intravenoso, el seguimiento del hiato osmolal puede ayudar a identificar a aquellos individuos en riesgo de desarrollar acidosis láctica, con valores superiores a 12 mg/dL, que sugieren un mayor riesgo de toxicidad por propilenglicol. El tratamiento incluye la suspensión del agente y, en casos graves, la eliminación mediante hemodiálisis. Teóricamente, el fomepizol, que inhibe la alcohol deshidrogenasa y ralentiza la descomposición de los alcoholes en sus metabolitos tóxicos, podría tener alguna utilidad, pero su eficacia en la intoxicación por propilenglicol no está clara.
Recuadro 2. Medicamentos de uso frecuente formulados con propilenglicol como vehículo
- Diazepam
- Esmolol
- Hidralazina
- Multivitamínicos
- Pentobarbital
- Fenitoína
- Digoxina
- Etomidato
- Lorazepam
- Nitroglicerina
- Fenobarbital
- Trimetoprim-sulfametoxazol
Inhibidores de la Transcriptasa Inversa Análogos de Nucleósidos/Nucleótidos
Los inhibidores de la transcriptasa inversa análogos de nucleósidos/nucleótidos (ITIAN) han revolucionado el tratamiento del VIH y el SIDA (Tabla 1). Un régimen típico de terapia antirretroviral altamente activa consiste en dos ITIAN y un inhibidor de la proteasa o un ITINN. Las toxicidades debidas a los ITIAN probablemente se deben a la toxicidad mitocondrial. Los ITIAN pueden inhibir la ADN polimerasa-γ, lo que interfiere con la síntesis de ADN mitocondrial y puede conducir a una transcripción y traducción anormales. Aunque actualmente no hay correlación clínica, los estudios demuestran que la zalcitabina tiene la mayor inhibición de la ADN polimerasa γ y que la lamivudina, el abacavir y el tenofovir tienen la menor. La oxidación de ácidos grasos de cadena larga también se ve afectada por los ITIAN. Uno de los resultados más graves de la toxicidad mitocondrial es la acidosis láctica. Las elevaciones del ácido láctico son comunes en individuos tratados con ITIAN, afectando aproximadamente al 9% de los individuos en un estudio. En este grupo, entre aquellos con lactato elevado, un poco más de un tercio tenía síntomas posiblemente consistentes con hiperlactatemia, incluyendo mialgias, fatiga y vómitos. La acidosis láctica puede ocurrir en regímenes con un solo ITIAN o con combinaciones de ITIAN. Los factores de riesgo incluyen la exposición a didanosina, estavudina o una combinación de ambas; sexo femenino; edad mayor de 40 años; inmunosupresión avanzada (recuentos de CD4 más bajos); y una duración más corta de la terapia (<12 meses), lo que sugiere una reacción idiosincrásica. En pacientes con síntomas de náuseas, vómitos, dolor abdominal, esteatosis hepática o transaminemia inexplicables, puede ser beneficioso verificar los niveles de lactato. La acidosis láctica generalmente se resuelve con la suspensión del ITIAN y terapia de apoyo, aunque algunos investigadores han sugerido la suplementación con riboflavina, tiamina, carnitina y coenzima Q-10. Un estudio que utilizó ensayo de PCR para comparar el ADN mitocondrial con el ADN nuclear en controles no infectados por VIH, individuos infectados por VIH que no tomaban ITIAN e individuos infectados por VIH que tomaban ITIAN con lactato elevado demostró que el VIH por sí solo puede afectar la relación ADN mitocondrial a nuclear y, por lo tanto, el VIH por sí solo podría resultar en acidosis láctica.
Metformina
Las biguanidas, metformina y fenformina, se han utilizado para tratar la diabetes mellitus desde la década de 1950. La fenformina fue retirada del mercado estadounidense en 1976 debido a más de 300 informes de acidosis láctica. La metformina, disponible desde 1995, tiene un riesgo mucho menor de acidosis láctica que su predecesora, la fenformina. La metformina inhibe la gluconeogénesis a partir del lactato en el hígado de manera dependiente del tiempo y la concentración.
En caso de sobredosis, la metformina se une a las membranas mitocondriales, específicamente al complejo I, lo que resulta en la inhibición del sistema de transporte de electrones, lo que lleva a un cambio hacia el metabolismo anaeróbico. La mayoría de los casos de acidosis láctica relacionada con metformina han sido en casos de sobredosis intencional o en individuos con condiciones subyacentes, como insuficiencia renal, insuficiencia cardíaca congestiva, insuficiencia hepática, sepsis y shock. El riesgo de muerte en estos pacientes se correlaciona mejor con la disfunción orgánica que con el nivel de lactato o el nivel de metformina. Hay una baja mortalidad en la sobredosis intencional de metformina con reconocimiento temprano, soporte hemodinámico y respiratorio y hemodiálisis. Con hemodiálisis, aproximadamente el 12% por hora del fármaco se elimina en las primeras 2 horas. Después de 15 horas de hemodiálisis, la eliminación es de aproximadamente 1.5% por hora.
Propofol
El síndrome de infusión de propofol es una complicación rara y potencialmente mortal de la infusión de propofol. Caracterizado por acidosis metabólica, rabdomiólisis del músculo esquelético y cardíaco, arritmias, insuficiencia miocárdica, insuficiencia renal, hepatomegalia y muerte, tiene un inicio repentino que generalmente resulta en la muerte. La mayoría de los informes de casos han sido en individuos que reciben dosis relativamente altas (>4 mg/kg/h) durante un período prolongado (>48 horas), pero también se ha descrito en infusiones de dosis bajas (1.4–2.6 mg/kg/h). El propofol puede alterar la cadena respiratoria mitocondrial de múltiples maneras. Se ha descrito una reducción en la actividad del citocromo-c oxidasa y una disminución de la actividad del complejo IV y una oxidación de ácidos grasos alterada con deterioro secundario de la actividad del complejo II en el síndrome de infusión de propofol. La nueva acidosis metabólica en un paciente con infusión de propofol debe hacer sospechar el síndrome de infusión de propofol. Se recomienda la suspensión del propofol, la reanimación y estabilización cardiocirculatoria y la hemodiálisis para eliminar el propofol; sin embargo, el síndrome es a menudo rápidamente fatal y los pacientes no responden a los vasopresores e inotrópicos.
Linezolid
La acidosis láctica relacionada con linezolid no se informó durante los ensayos clínicos de fase III, pero han surgido muchos informes de casos y series de casos desde entonces. La mayoría de los casos ocurren después de una duración prolongada de la terapia,pero hay informes con una duración de la terapia tan corta como 7 días. En la mayoría de los casos, la suspensión del linezolid resulta en la resolución de la acidosis láctica. El linezolid mata las bacterias uniéndose al ARN ribosómico (ARNr) 23S de la bacteria. En mamíferos, la subunidad ribosomal grande 16S del ARN es homóloga al ARNr 23S bacteriano. En dos pacientes con acidosis láctica relacionada con linezolid, se identificaron polimorfismos en el ARNr 16S mitocondrial, específicamente en la región estructuralmente similar al sitio de unión del linezolid ubicado en el ARNr 23S bacteriano. Se debe tener precaución al administrar linezolid con inhibidores selectivos de la recaptación de serotonina, ya que bloquean la actividad de la glicoproteína p y pueden elevar los niveles de linezolid. Se ha informado síndrome serotoninérgico con el uso concomitante de linezolid e inhibidores selectivos de la recaptación de serotonina.
Agentes B2-Adrenérgicos: Epinefrina, Ritodrina, Terbutalina, Salbutamol y Dobutamina
Los agonistas beta causan acidosis láctica de varias maneras. En primer lugar, la estimulación Muscular y hepática mediada por receptores b2-adrenérgicos de la fosforilasa y la inhibición de la glucógeno sintetasa estimulan la glucólisis y, por lo tanto, un aumento en la producción de piruvato. En el músculo esquelético, los agonistas beta estimulan la Na+-K+-ATPasa a través de la regulación positiva del AMPc. Esto aumenta la generación de ADP y luego de fosfofructoquinasa, lo que acelera la glucólisis. Luego, los agonistas beta inhiben la PDH, lo que conduce a una disminución de la oxidación del piruvato a acetil CoA y, por lo tanto, a un aumento de la reducción del piruvato a lactato.
Salicilatos
La sobredosis de salicilato se caracteriza por una alcalosis respiratoria temprana y una acidosis metabólica tardía, debida principalmente a la acidosis láctica. La acidosis respiratoria ocurre tarde y es típicamente un evento terminal. La toxicidad de los salicilatos se debe a múltiples mecanismos, incluyendo la inhibición de la beta-oxidación de ácidos grasos, la disminución de la disponibilidad de CoA, la inhibición de la succinato deshidrogenasa y la alfa-cetoglutarato deshidrogenasa, y el aumento de la permeabilidad de la membrana mitocondrial interna. Además, la alcalosis respiratoria temprana estimula un aumento en el nivel de 2,3-difosfoglicerato, con el consiguiente aumento de la actividad de la fosfofructoquinasa, lo que resulta en una glucólisis acelerada. El tratamiento consiste en administrar glucosa para evitar la hipoglucemia del sistema nervioso central y mantener el pH entre 7.45 y 7.5 para disminuir la concentración de salicilato en el sistema nervioso central y aumentar la excreción renal de salicilato. La hemodiálisis está indicada en intoxicaciones graves.
Sulfasalazina
La sulfasalazina se descompone en el colon en sulfapiridina y ácido 5-aminosalicílico (5-ASA). La sulfapiridina se absorbe y se acetila principalmente en el hígado, y el resto se somete a glucuronidación o hidroxilación antes de ser excretada en la orina. Aproximadamente una cuarta parte del 5-ASA se absorbe, que luego se acetila a N-acetil-5-ASA. En un informe de caso, la acidosis láctica con coingestión de paracetamol resultó en niveles de lactato de aproximadamente 20mmol/L con supervivencia del paciente a pesar de la administración tardía de N-acetilcisteína.129
Otros Medicamentos que Causan Hiperlactatemia
Se han publicado informes de casos de acidosis láctica debida a isoniazida (INH), simvastatina, atorvastatina, niacina, y ácido nalidíxico. No está claro si la hiperlactatemia se debe a la insuficiencia hepática que ocurre con estos fármacos o si hay una inhibición adicional de la respiración celular. La INH inhibe el piridoxal micobacteriano (la forma oxidada de la piridoxina) para bloquear el crecimiento y el metabolismo bacterianos. Los niveles elevados de INH podrían afectar los dinucleótidos de difosfopiridina humanos y disminuir el metabolismo del lactato a piruvato. La simvastatina y la atorvastatina pertenecen a una clase de fármacos conocidos por reducir el coenzima Q10 sérico, un cofactor central de la cadena respiratoria mitocondrial. La lactulosa ha sido un pilar del tratamiento de la encefalopatía hepática durante décadas. Las bacterias colónicas descomponen la lactulosa en ácidos láctico, acético y fórmico. En un caso en el que un paciente cirrótico recibió tratamiento con lactulosa, se produjo una acidosis láctica grave, teóricamente debido a la formación de lactato en el colon, que se difundió de regreso a través de la pared intestinal, facilitada por la hipomotilidad intestinal.
Tiamina, Biotina y Hierro
La deficiencia de tiamina que resulta en acidosis láctica se describe con mayor frecuencia en pacientes con alcoholismo, pacientes que reciben nutrición parenteral total, trabajadores extranjeros cuyos hábitos nutricionales han cambiado, y lactantes que reciben una fórmula defectuosa a base de soja. Se registraron niveles de lactato de hasta 20mmol/L en un estudio. La tiamina, una vitamina hidrosoluble del complejo B que los mamíferos no pueden sintetizar, es un cofactor esencial para las enzimas del citosol y las mitocondrias, incluyendo la cetolasa, la PDH y la alfa-cetoglutarato deshidrogenasa. Se cree que las pérdidas en la actividad de la PDH y la alfa-cetoglutarato deshidrogenasa conducen a una reducción de la entrada de piruvato en el ciclo del TCA y a un aumento de la producción de lactato. La falta de transcetolasa conduce a una reducción de la vía de las pentosas fosfato y la consiguiente reducción de NADH, estimulando la glucólisis anaeróbica y una mayor producción de lactato. La administración de glucosa estimula la producción excesiva de lactato.
La biotina es un cofactor en múltiples reacciones de carboxilación, incluyendo la carboxilación del piruvato para formar oxaloacetato. La deficiencia de biotina se demostró en humanos alimentándolos con una dieta de claras de huevo crudas. Las claras de huevo crudas tienen una glicoproteína llamada avidina, que se une a la biotina. Cocinar las claras desnaturaliza la avidina y destruye su afinidad por la biotina. En individuos alimentados con una dieta de clara de huevo, se observaron síntomas de dermatitis, palidez, cambios en el estado mental, mialgias, anorexia, anemia y cambios en el ECG consistentes con isquemia coronaria. La administración de biotina inyectable resolvió todos los síntomas en un plazo de 3 a 5 días. La deficiencia de biotina también se ha descrito en individuos que reciben nutrición parenteral total sin biotina; con el uso prolongado de algunos anticonvulsivantes, como fenitoína, carbamazepina, fenobarbital y primidona; y en mutaciones genéticas. Teóricamente, el uso prolongado de antibióticos podría disminuir los niveles de biotina, ya que la biotina es generada por bacterias que viven en el intestino, y existen receptores especializados en el colon para la absorción de biotina; sin embargo, faltan datos primarios al respecto.
El hierro es esencial para la respiración celular, ya que desempeña un papel importante en el transporte de electrones a través de los centros de hierro-azufre y de los citocromos a, b y c. Los estados de sobrecarga de hierro y deficiencia de hierro se han asociado con acidosis láctica. Un síndrome conocido como GRACILE lleva el nombre de los hallazgos clínicos de retraso del crecimiento, aminoaciduria, colestasis, sobrecarga de hierro, acidosis láctica y muerte temprana.
Alcoholes de Azúcar (Fructosa, Sorbitol y Xilitol)
La fructosa entra en la vía glucolítica como lactato, glucosa o glucógeno. El sorbitol se absorbe mal en el intestino y se convierte principalmente en fructosa, y solo una pequeña parte se metaboliza a glucosa. El xilitol también se absorbe mal cuando se administra por vía oral, pero aumenta con el tiempo, debido a adaptaciones en la flora oral. El xilitol se metaboliza a glucosa, glucógeno, lactato y xilulosa. Se ha informado acidosis láctica cuando se utilizó fructosa, sorbitol, o xilitol en nutrición parenteral. Se ha demostrado toxicidad letal por xilitol caracterizada por aumento de la osmolalidad sérica, aumento de la producción de orina y hemoconcentración en animales.
Estrícnina
La malaria ha sido una de las principales causas de muerte por enfermedad, particularmente en África subsahariana, durante siglos. La acidosis láctica se utiliza como indicador pronóstico en la infección malárica aguda. La etiología de la elevación del lactato no está clara, pero puede estar relacionada con la disminución del flujo sanguíneo hepático y la disminución de la depuración de lactato, la producción de lactato por los parásitos en los glóbulos rojos a través de la glucólisis anaeróbica, o un desajuste entre VO2 y la demanda de oxígeno. La administración de N-acetilcisteína a adultos con malaria grave resultó en una rápida disminución del lactato, pero se desconoce si mejoraría la mortalidad.
Ácido Valproico
El ácido valproico inhibe la fosforilación oxidativa disminuyendo la actividad del citocromo aa3 y del citocromo c, inhibiendo el transporte de succinato, y cambiando la conformación de las proteínas de la membrana mitocondrial. Se han asociado informes de acidosis láctica con el uso de ácido valproico en individuos con anomalías mitocondriales subyacentes y en sobredosis de valproato. La retirada del medicamento, la hemodiálisis, y los cuidados de apoyo dieron como resultado un buen resultado.
Cocaína y Metanfetamina
A mediados de la década de 1980, dos atletas de élite, Len Bias y Don Rogers, murieron por consumo de cocaína. La muerte por cocaína generalmente se debe a convulsiones, acidosis láctica, hipertermia, infartos de miocardio y arritmias. La cocaína estimula la actividad adrenérgica, lo que lleva a un aumento de la glucólisis y la producción de ácido láctico. Han surgido informes de acidosis láctica relacionada con metanfetamina. La intoxicación aguda por metanfetamina puede presentarse con coma, shock, convulsiones, hipertermia, insuficiencia renal y acidosis.
La metanfetamina disminuye la actividad del complejo I en las mitocondrias. También induce una disminución de la proliferación, un aumento de la apoptosis y la alteración de las redes mitocondriales, lo que resulta en fragmentación mitocondrial.
Compuestos Cianogénicos: Cianuro, Nitrilos Alifáticos y Nitroprusiato
El cianuro o los productos químicos que liberan cianuro se encuentran en el aislamiento; pesticidas; soluciones de decapado y galvanoplastia de metales; los huesos de albaricoques, cerezas y melocotones; y raíces de mandioca, almendras y brotes de bambú.Los nitrilos alifáticos se utilizan en la fabricación de fibras sintéticas, resinas, plásticos, productos farmacéuticos y vitaminas. Los dos nitrilos más estudiados, el metacrilonitrilo y el acrilonitrilo, liberan cianuro durante el metabolismo de estas sustancias a través de la epoxidación en el hígado. El nitroprusiato de sodio es un medicamento utilizado por su efecto vasodilatador. Está compuesto por una molécula de hierro unida a cinco moléculas de cianuro y una molécula de óxido nítrico. El riesgo de toxicidad por cianuro por la infusión de nitroprusiato aumenta con dosis altas durante un período prolongado. Este riesgo también es mayor cuando se administra a individuos con desnutrición o insuficiencia renal preexistente. El cianuro se une al citocromo inhibiendo la actividad del citocromo-c oxidasa. Esta inhibición del metabolismo oxidativo resulta en una disminución de la extracción tisular de oxígeno y un cambio del metabolismo celular hacia la glucólisis anaeróbica. Una disminución en la diferencia de oxígeno arteriovenoso debe aumentar la sospecha de intoxicación por cianuro. El tratamiento de la sospecha de toxicidad por cianuro implica cuidados de apoyo y tratamiento con un agente formador de metahemoglobina y posteriormente un donante de azufre. El nitrito de amilo o el nitrito de sodio forman metahemoglobina, que tiene una alta afinidad por el cianuro, disociándolo del citocromo oxidasa y restaurando así la fosforilación oxidativa. Luego se administra tiosulfato de sodio como sustrato donante de azufre para la conversión del cianuro en una forma menos tóxica, el tiocianato, que se excreta renalmente. La diálisis, en particular la hemodiafiltración venovenosa continua, también puede ser útil como medida adicional en intoxicaciones graves. En el caso de toxicidad por cianuro inducida por nitroprusiato, la adición de hidroxocobalamina puede disminuir la toxicidad.
Anestésicos Generales: Éter Dietílico y Halotano
El éter dietílico se utiliza comúnmente como solvente de laboratorio y se utiliza en la producción de plásticos de celulosa. Se utilizó como anestésico general hasta que se dispuso de anestésicos inhalatorios no inflamables y menos tóxicos, como el halotano. Se informa que el éter dietílico y el halotano son agentes causantes de acidosis láctica, pero los mecanismos no se han descrito bien. Se ha demostrado que el éter dietílico, los derivados del éter dietílico y el halotano alteran la fosforilación oxidativa.
5-Fluorouracilo
Los informes de acidosis láctica, hiperamonemia y encefalopatía con infusiones de 5-fluorouracilo (5-FU) son raros. La administración de 5-FU en individuos con deficiencias congénitas de dihidropirimidina deshidrogenasa y dihidropirimidina es letal. En micrografías electrónicas, se observa degradación y alteración en la estructura mitocondrial y probablemente está relacionada con la inhibición de la síntesis de ADN y ARN por parte del 5-FU.
Tipo B3 – Errores innatos del metabolismo
Existen muchas anomalías genéticas que se sabe que causan síndromes de acidosis láctica. Las afecciones que resultan en deficiencia en la fosforilación oxidativa, como el síndrome de Kearns-Sayre; el síndrome de Pearson; MERRF; y el síndrome de encefalomiopatía mitocondrial, acidosis láctica y accidente cerebrovascular (MELAS), son encefalomiopatías mitocondriales comúnmente asociadas con acidosis láctica. El manejo de estos individuos es típicamente de apoyo. Un ensayo que intentó usar DCA para reducir el ácido láctico en pacientes con MELAS resultó en neuropatía periférica y se suspendió. La deficiencia de fructosa-1,6-difosfatasa provoca hipoglucemia potencialmente mortal y acidemia láctica solo cuando ocurre el ayuno, ya que la gluconeogénesis está alterada. Esto es más pronunciado en la enfermedad de almacenamiento de glucógeno tipo I, también conocida como enfermedad de von Gierke. Una deficiencia de glucosa-6-fosfatasa, translocasa de glucosa-6-fosfato o la translocasa de fosfato del retículo endoplásmico resulta en una glucogenólisis y gluconeogénesis comprometidas. La deficiencia de PDH puede deberse a varias mutaciones con una gradación en el fenotipo. La alteración puede variar desde acidosis láctica infantil fatal hasta ataxia como la alteración principal. La deficiencia de piruvato carboxilasa también tiene diferentes expresiones fenotípicas dependiendo del grado de alteración. También se caracteriza por hipoglucemia, acidosis láctica y cetosis. El fenotipo tipo C carece del retraso psicomotor observado en el tipo A. A diferencia del tipo B, donde los lactantes típicamente mueren a los 3 meses de edad, estos individuos sobreviven hasta la edad adulta con episodios de acidemia láctica. La aciduria metilmalónica es causada por una deficiencia de metilmalonil-CoA mutasa o por defectos en el transporte, captación o síntesis de 5′-desoxiadenosilcobalamina. La presentación clínica varía, pero puede incluir acidosis láctica, hipoglucemia, cetosis e hiperamonemia. Se ha utilizado diálisis para eliminar la acidemia durante las crisis metabólicas, pero ha habido poco éxito para reducir el daño orgánico terminal, que resulta de la acumulación de los ácidos orgánicos tóxicos.
Acidosis D-láctica
El D-lactato es el isómero óptico del L-lactato. Los niveles elevados de D-lactato generalmente se deben a la producción bacteriana en el tracto intestinal, aunque también pueden deberse a la ingestión de D-lactato, a la producción endógena a través de la vía de la metilglioxalasa o a la infusión de soluciones de DL-lactato en diálisis peritoneal. La presentación clínica es única por sus manifestaciones neurológicas inusuales, que van desde el habla arrastrada hasta el comportamiento abusivo que duran desde unas pocas horas hasta varios días. Se ha demostrado que las comidas ricas en glucosa exacerban los síntomas. También está presente una acidosis metabólica grave, pero los niveles de L-lactato son normales. Se debe solicitar D-lactato para hacer el diagnóstico. La mayoría de los pacientes que presentan acidosis D-láctica tienen síndrome de intestino corto. Se teoriza que los síntomas neurológicos se manifiestan como resultado de la toxicidad del D-lactato para el cerebro, que carece de D-2-hidroxiácido deshidrogenasa, la enzima que convierte el ácido D-láctico en piruvato. El tratamiento de esta afección incluye una dieta baja en carbohidratos, enema de solución salina en caso de estreñimiento y antibióticos orales poco absorbibles en un esfuerzo por disminuir las bacterias intestinales productoras de D-lactato. Durante los episodios de acidosis, la administración de insulina puede disminuir los niveles de ácidos grasos, lo que puede resultar en un aumento de la oxidación y eliminación del D-lactato. En casos graves, se puede utilizar hemodiálisis.