El desarrollo de insuficiencia cardíaca en el contexto de estrés crónico, como hipertensión o infarto de miocardio (IM), se caracteriza inicialmente por cambios complejos en la estructura y función del corazón a nivel molecular, celular y orgánico. Este proceso dinámico, denominado remodelación cardíaca, conduce a disfunción contráctil, dilatación de las cavidades, disincronía ventricular y arritmias. A nivel celular, el corazón remodelado manifiesta alteraciones significativas tanto en los miocitos cardíacos como en células no miocárdicas, como fibroblastos, células endoteliales y células inmunitarias. Además, se producen cambios significativos en la vasculatura miocárdica y en la composición de la matriz extracelular.
Los cambios progresivos en el fenotipo del miocito cardíaco constituyen una anomalía central en el corazón sometido a estrés crónico y con insuficiencia cardíaca. El fenotipo comprende múltiples componentes, incluyendo hipertrofia celular y alteraciones en el manejo del calcio, la función sarcomérica, las propiedades eléctricas, la homeostasis redox, el metabolismo, la energética y la viabilidad celular, que en conjunto contribuyen significativamente al fenotipo cardíaco global. La hipertrofia del miocito también se observa en condiciones fisiológicas (p. ej., embarazo o en atletas), pero en este caso no se acompaña de cambios perjudiciales como el deterioro de la contractilidad. Esta divergencia en los fenotipos indica que los diferentes componentes del fenotipo del miocito pueden regularse de forma independiente, al menos hasta cierto punto. Incluso en un contexto de enfermedad (p. ej., al inicio de una sobrecarga de presión), el corazón puede hipertrofiarse pero mantener la función contráctil (remodelación adaptativa), mientras que con un estrés crónico más intenso comienza a fallar. Por lo tanto, el fenotipo general puede estar determinado por el equilibrio entre los procesos potencialmente adaptativos y desadaptativos que ocurren dentro del miocito cardíaco.
En este capítulo, revisamos las principales alteraciones celulares de los cardiomiocitos que contribuyen a la patogénesis de la insuficiencia cardíaca. Comenzamos analizando la base celular de la disfunción contráctil, la manifestación cardíaca clave de la insuficiencia cardíaca. Se considera la contribución de los cambios en el acoplamiento excitación-contracción (en particular, el manejo del calcio y la función sarcomérica) a la disfunción contráctil y la arritmogénesis. A continuación, analizamos diversas alteraciones globales dentro de los cardiomiocitos que también afectan la función contráctil y la viabilidad celular, como los cambios en la homeostasis redox y la señalización, las respuestas al estrés celular y el recambio macromolecular y proteico. Como se verá, estos procesos globales interactúan entre sí y tienen efectos complejos en el proceso de remodelación (por ejemplo, la homeostasis redox y la señalización modulan el acoplamiento excitación-contracción, así como las respuestas al estrés). El fenotipo del cardiomiocito en el corazón con insuficiencia cardíaca también puede verse afectado por otros tipos de células cardíacas y, a su vez, influir en estos tipos celulares. Por lo tanto, se analiza el papel de las interacciones de los cardiomiocitos con otros tipos celulares, en particular fibroblastos, células endoteliales y células inmunitarias/inflamatorias. Finalmente, se revisa el papel de la regulación por ARNm no codificantes, como los microARN (miARN), que a menudo tienen efectos globales en las vías de señalización celular en el corazón con insuficiencia cardíaca. Las vías de señalización que subyacen al desarrollo de la hipertrofia de los miocitos se abordan en otra sección de esta edición. Asimismo, el importante papel de las alteraciones en la energética y el metabolismo miocárdicos, que, por ejemplo, pueden tener un gran impacto en la función contráctil durante la insuficiencia cardíaca, se trata en otros capítulos.
Disfunción contráctil
Los miocitos cardíacos en el corazón con insuficiencia cardíaca presentan diversas anomalías en la función contráctil, incluyendo una reducción en la amplitud y la fuerza de contracción, una ralentización de la contracción y la relajación, un aumento de la fuerza diastólica y respuestas alteradas a los cambios en la frecuencia cardíaca y la estimulación β-adrenérgica. Las alteraciones tanto en el acoplamiento excitación-contracción como en las propiedades del sarcómero contribuyen a estas anomalías. Ofrecemos una breve descripción general del acoplamiento excitación-contracción y la función sarcomérica normales, y luego abordamos las anomalías específicas que se presentan en la insuficiencia cardíaca.
Acoplamiento excitación-contracción normal
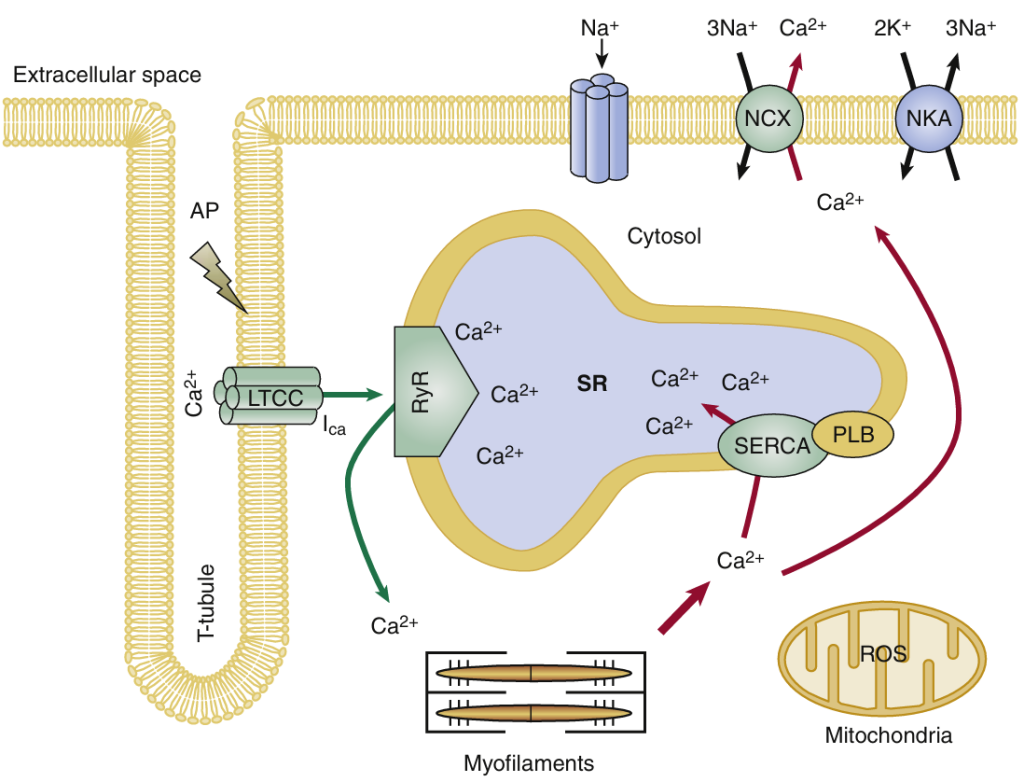
La función cardíaca fisiológica requiere la activación coordinada, tanto temporal como espacial, del corazón. A nivel celular, este proceso finamente ajustado está regulado principalmente por flujos de Ca2+ sincronizados con precisión en cada cardiomiocito (Fig. 1). Cuando un potencial de acción despolariza la célula, los canales de Ca2+ de tipo L dependientes de voltaje (LTCC), ubicados principalmente en los túbulos transversos (túbulos T), se abren para generar una corriente de Ca2+ de entrada (ICa), que induce un aumento localizado de Ca2+ en la hendidura diádica, cerca de los canales de liberación de Ca2+ o del receptor de rianodina (RyR2) del retículo sarcoplásmico (RS). Este influjo transarcolémico de Ca2+ activa el RyR2 y da lugar a la denominada liberación de Ca2+ inducida por Ca2+ desde el RS, que constituye el componente principal del aumento de Ca2+ citosólico durante la sístole (es decir, el transitorio de Ca2+). La concentración intracelular de Ca2+ aumenta de aproximadamente 100 nmol/L durante la diástole a aproximadamente 1 μmol/L durante la sístole, lo que provoca la activación y contracción de los miofilamentos. La repolarización del potencial de membrana se induce por la inactivación de ICa y la activación de las corrientes de K+ rectificadoras retardadas. Durante la diástole, el Ca2+ se elimina del citosol a través de dos vías principales: (1) la ATPasa de Ca2+ del retículo sarcoplásmico (SERCA2a), ubicada en la membrana del retículo sarcoplásmico, que bombea Ca2+ de vuelta al lumen del retículo sarcoplásmico; (2) el intercambiador de Na+/Ca2+ del sarcolema (NCX1), que transfiere Ca2+ al espacio extracelular. Mediante estos dos mecanismos de transporte de Ca2+, la concentración intracelular de Ca2+ ([Ca2+]i) disminuye hasta alcanzar concentraciones fisiológicas de reposo de aproximadamente 100 nmol/L, lo que permite que la célula se relaje y recupere su longitud de reposo diastólica fisiológica.
Alteración en el manejo del Ca2+ en miocitos cardíacos con insuficiencia cardíaca
La prolongación de la duración del potencial de acción, la disminución de la capacidad generadora de fuerza y la ralentización de las tasas de contracción y relajación son los cambios funcionales característicos del corazón humano con insuficiencia cardíaca. La alteración del manejo del Ca2+ es una característica clave del miocito cardíaco con insuficiencia cardíaca, con gran relevancia fisiopatológica para el deterioro progresivo de la función contráctil del corazón. Las alteraciones específicas en los niveles de expresión, así como las modificaciones postraduccionales de importantes proteínas cardíacas implicadas en el manejo del Ca2+, contribuyen causalmente a la disfunción contráctil sistólica y diastólica y a una mayor propensión a las arritmias cardíacas. Las modificaciones postraduccionales que alteran la función de las proteínas clave implicadas en el manejo del Ca2+ incluyen alteraciones en la fosforilación, la nitrosilación, el estado de oxidación y la sumoilación. La fosforilación alterada de proteínas se produce como consecuencia de cambios en la actividad de diversas quinasas (por ejemplo, la proteína quinasa dependiente de AMPc [PKA], la quinasa II dependiente de calcio-calmodulina [CaMKII]), así como de perturbaciones en la actividad de la fosfatasa (por ejemplo, la proteína fosfatasa 1 [PP1]).
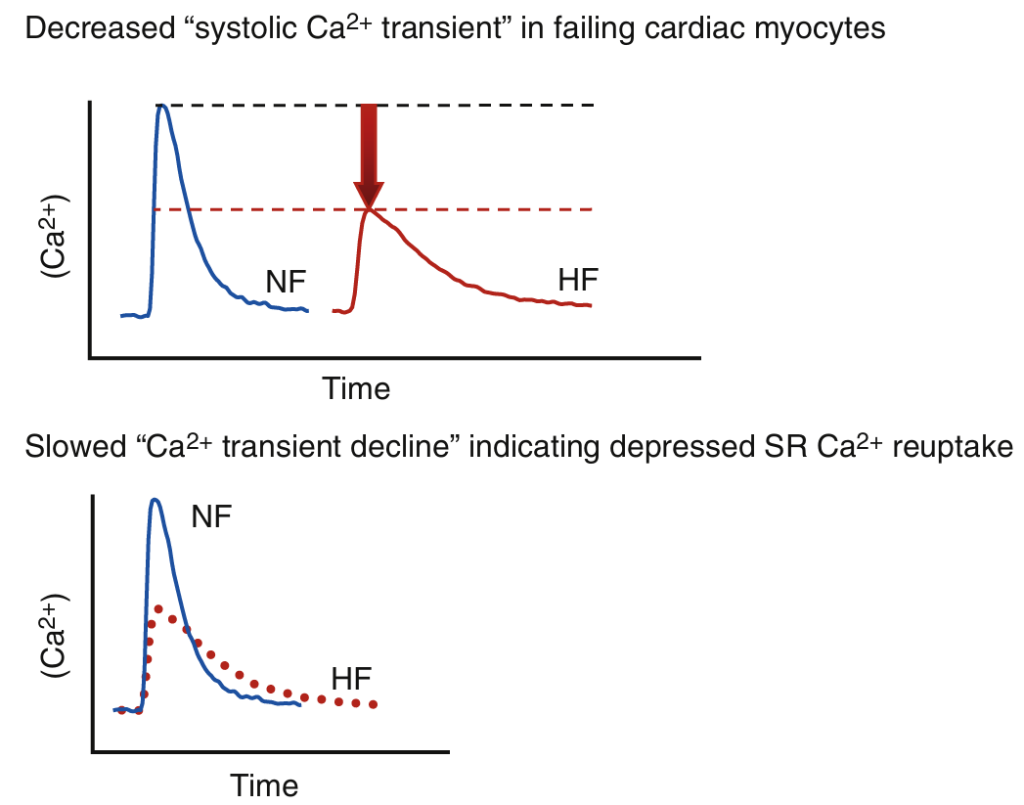
El miocito cardíaco con insuficiencia cardíaca presenta una amplitud significativamente reducida del transitorio sistólico de Ca2+ en comparación con los miocitos de control sanos (Fig. 2), lo que constituye un factor importante responsable de la reducción de la amplitud contráctil de la célula con insuficiencia. Los miocitos con insuficiencia cardíaca también suelen presentar una disminución más lenta del transitorio de Ca2+ durante la diástole, lo que contribuye significativamente a la relajación anormal (retrasada). Además, el aumento normal de la amplitud del transitorio de Ca2+ (y, por lo tanto, de la fuerza de contracción) que se produce con una frecuencia cardíaca más rápida se ve atenuado o incluso invertido en el corazón con insuficiencia cardíaca (es decir, la relación fuerza-frecuencia [RFF] positiva normal se convierte en una RFF plana o negativa). En general, se acepta que una reducción en el contenido de Ca2+ del retículo sarcoplásmico es una de las principales razones de la disminución de la amplitud del transitorio sistólico de Ca2+ y de la RFF anormal. Se ha observado consistentemente una disminución del contenido de Ca2+ en el retículo sarcoplásmico (RS) en miocitos aislados de corazones humanos y animales con insuficiencia cardíaca, mientras que las alteraciones en la entrada de Ca2+ mediada por LTTC parecen ser menos relevantes. Desde un punto de vista mecanicista, una reducción en el contenido de Ca2+ del RS puede deberse a una recarga diastólica insuficiente de Ca2+ (o carga) del RS o a una mayor pérdida de Ca2+ a través de los canales de liberación de Ca2+ RyR2 durante la diástole, y también puede verse influenciada por cambios en la actividad de NCX. De hecho, los tres mecanismos pueden contribuir a la reducción del contenido de Ca2+ del RS y al fenotipo contráctil del miocito con insuficiencia cardíaca (Fig. 3).
Recaptación reducida de Ca2+ por el retículo sarcoplásmico en la insuficiencia cardíaca
Durante la diástole, la SERCA2a bombea Ca2+ al lumen del retículo sarcoplásmico (RS) y proporciona un contenido suficiente de Ca2+ para su liberación durante la sístole posterior. La captación diastólica de Ca2+ en el RS, dependiente de la SERCA2a, normalmente predomina sobre la extrusión transarcolémica de Ca2+ a través del intercambiador Na+/Ca2+ (NCX). La SERCA2a está regulada por la fosfoproteína fosfolambano (PLB), que, tras su fosforilación por la CaMKII (en Thr17) y/o la PKA (en Ser-16), libera su efecto inhibidor sobre la SERCA2a debido a la disociación del complejo PLB/SERCA2a. Los niveles de proteína SERCA2a se reducen en el miocardio con insuficiencia cardíaca, lo que se acompaña de una reducción en la sumoilación de la SERCA2a y provoca una alteración en la recaptación diastólica de Ca2+ en el RS. Además, los niveles de PLB permanecen inalterados y su estado de fosforilación puede verse reducido, lo que provoca una mayor inhibición relativa de SERCA2a por la PLB no fosforilada, agravando así el deterioro de la recaptación de Ca2+ (véase la figura 3). La disminución del contenido de Ca2+ en el retículo sarcoplásmico reduce la disponibilidad de Ca2+ para el transitorio sistólico de Ca2+ subsiguiente y perjudica la función sistólica.
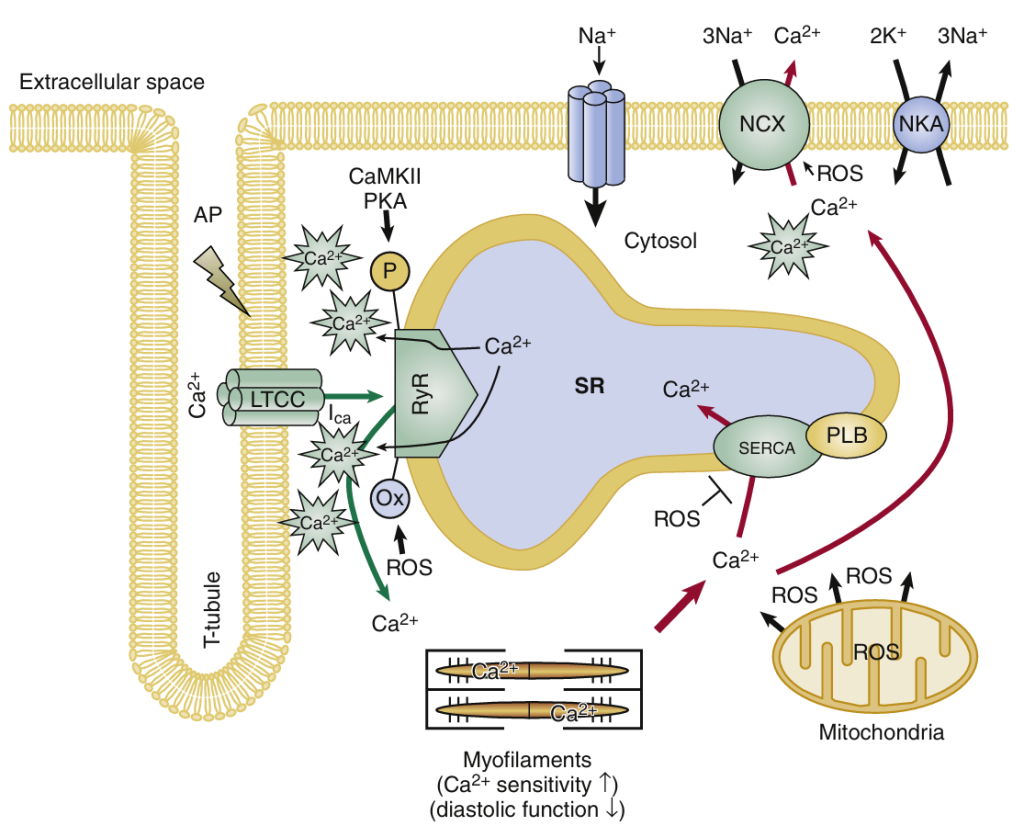
La disminución de la expresión de SERCA2a también puede afectar la función diastólica del corazón con insuficiencia cardíaca. Si ningún otro mecanismo (como un aumento de la actividad de NCX) compensa la reducción de la función de SERCA2a con respecto a la eliminación de Ca2+ del citosol durante la diástole, entonces se produce una sobrecarga de Ca2+ citosólico diastólico. Los miocitos con niveles elevados de Ca2+ diastólico presentarán una activación persistente de bajo nivel de los miofilamentos en un momento en que estos deberían estar completamente relajados, lo que resulta en un aumento de la fuerza diastólica y disfunción diastólica. Esta incapacidad para relajarse completamente durante la diástole dificulta el llenado del corazón y, por lo tanto, también puede empeorar la disfunción sistólica. Además, los niveles anormalmente elevados de Ca2+ diastólico pueden tener otros múltiples efectos, como cambios en la transcripción genética, la viabilidad celular y la función mitocondrial a través de la activación alterada de quinasas dependientes de Ca2+ (por ejemplo, CaMKII), fosfatasas (por ejemplo, calcineurina), enzimas mitocondriales, caspasas y otros mecanismos.
En vista de estos efectos sobre la función contráctil, así como de otros aspectos del fenotipo de la insuficiencia cardíaca, restaurar la función de SERCA2a en la insuficiencia cardíaca podría representar un enfoque terapéutico prometedor. Estudios experimentales demostraron que la sobreexpresión adenoviral de SERCA2a en cardiomiocitos humanos puede mejorar la contractilidad cardíaca, ya que restaura el contenido de Ca2+ del retículo sarcoplásmico y el transitorio sistólico de Ca2+, mientras que la reducción de los niveles de Ca2+ citosólico preserva la función diastólica. Además, se demostró que la sobreexpresión de SERCA2a mejora la energética miocárdica y la función endotelial, y que tiene efectos antiarrítmicos. La relevancia clínica potencial de la estimulación de SERCA2a se abordó en los ensayos CUPID (Regulación al alza del calcio mediante la administración percutánea de terapia génica en la enfermedad cardíaca), en los que pacientes con insuficiencia cardíaca fueron tratados con una única infusión de un vector viral adenoasociado (AAV) que administraba SERCA2a frente a placebo. Si bien este estudio inicial sugirió una posible eficacia, el estudio más amplio, el ensayo CUPID2, no logró demostrar eficacia en la insuficiencia cardíaca avanzada.
Aumento de la actividad de NCX en el corazón con insuficiencia cardíaca
El NCX se localiza en el sarcolema del cardiomiocito, donde, en su modo de conducción, transfiere un ion Ca2+ al espacio extracelular a cambio de tres iones Na+, aprovechando el gradiente transmembrana de Na+ (véase la figura 1). Este mecanismo es electrogénico, ya que produce el desplazamiento neto de una carga positiva hacia el citosol y, por lo tanto, puede despolarizar el potencial de membrana e incluso tener efectos arritmogénicos en condiciones de aumentos espontáneos y localizados de [Ca2+]i. Se ha observado un aumento en la expresión y actividad del NCX en la insuficiencia cardíaca humana y experimental, lo que puede tener efectos funcionales complejos dependiendo del modo de actividad del NCX y la etapa de la insuficiencia cardíaca. Ante la disminución de la función de SERCA2a, el aumento de la actividad del NCX compite con SERCA2a por la eliminación de Ca2+ durante la diástole. Esto puede agravar aún más la disminución del contenido de Ca2+ en el retículo sarcoplásmico (RS), ya que hay menos Ca2+ citosólico disponible para la carga de Ca2+ en el RS mediada por SERCA2a. Sin embargo, el aumento de la función de NCX también puede proteger parcialmente a los miocitos cardíacos contra la sobrecarga diastólica grave de Ca2+ y la disfunción diastólica. De hecho, se encontró que un aumento en los niveles de NCX en muestras de miocardio humano explantadas se correlacionaba con la preservación de la función diastólica, mientras que los pacientes con disfunción diastólica tenían niveles reducidos de NCX. Por otro lado, la actividad de NCX puede contribuir a la sobrecarga de Ca2+ en situaciones donde hay sobrecarga intracelular de Na+. La razón es que a altas concentraciones intracelulares de [Na+]i, NCX cambia a un «modo inverso» y bombea Na+ a cambio de Ca2+. La mayor contribución de la entrada de Ca2+ dependiente de NCX, en contraposición a la liberación de Ca2+ del retículo sarcoplásmico durante la sístole en miocitos con insuficiencia, tiene efectos adversos sobre la captación mitocondrial de Ca2+ (que depende de altos gradientes de Ca2+) y promueve un aumento en los niveles de especies reactivas de oxígeno (ROS) mitocondriales debido a la actividad reducida de las deshidrogenasas del ciclo de Krebs dependientes de Ca2+ que normalmente mantienen las reservas antioxidantes. Este mecanismo perjudicial puede agravarse aún más de forma dependiente de las especies reactivas de oxígeno (ROS) y conducir a un círculo vicioso de alteración de los flujos de Ca2+ citosólicos y mitocondriales y un aumento del estrés oxidativo, ya que las ROS inducen una mayor sobrecarga de Na+ citosólico.
La fuga del receptor RyR2 provoca una pérdida diastólica de Ca2+ en el retículo sarcoplásmico en la insuficiencia cardíaca.
La fuga diastólica de Ca2+ del retículo sarcoplásmico (RS) debida a un aumento patológico de la probabilidad de apertura del RyR2 es un mecanismo importante que contribuye a la disminución del contenido de Ca2+ del RS en la insuficiencia cardíaca. El Ca2+ se escapa del RS mediante eventos de liberación espontánea y descoordinada de Ca2+ o «chispas de Ca2+». La expresión del RyR2 parece no modificarse en la insuficiencia cardíaca, pero su regulación funcional se ve alterada drásticamente por complejas modificaciones postraduccionales. Estas alteraciones implican un aumento en la fosforilación de RyR2 como resultado de quinasas de proteínas hiperactivas, como CaMKII15 y PKA,16 y posiblemente una reducción de la desfosforilación de RyR2.17 Un aumento en la oxidación o nitrosilación de RyR2 como consecuencia del aumento del estrés oxidativo y nitrosativo en la insuficiencia cardíaca también puede ser importante.5 Aunque la regulación transitoria dependiente de la fosforilación y redox de la activación de RyR2 puede cumplir funciones fisiológicas en miocitos sanos, en el miocito con insuficiencia la hiperfosforilación y/o oxidación de RyR2 conduce a una fuga diastólica severa de Ca2+ del SR. Además, el acoplamiento de LTCC a RyR también está afectado en la insuficiencia cardíaca debido a la remodelación de los túbulos T, de modo que algunos RyR están «huérfanos» y contribuyen a la liberación disincrónica de Ca2+.
Es importante dilucidar los mecanismos precisos de la hiperfosforilación de RyR2 y las quinasas responsables de esta anomalía, ya que podrían representar dianas terapéuticas, aunque siguen siendo objeto de debate. Si bien un laboratorio reportó evidencia sólida de una desregulación de RyR2 mediada por PKA en la insuficiencia cardíaca, otros no lograron demostrar un aumento en la hiperfosforilación dependiente de PKA. También existe evidencia de la participación de la fosforilación de RyR2 dependiente de CaMKII en la inducción de la fuga de Ca2+ del retículo sarcoplásmico. La desregulación de la apertura de RyR2 relacionada con el estado redox en el corazón con insuficiencia cardíaca⁵ podría implicar un aumento de las especies reactivas de oxígeno (ROS) producidas por las mitocondrias u otras fuentes, como las NADPH oxidasas, y está relacionada con la oxidación de residuos de cisteína específicos dentro de RyR2. Curiosamente, ambas quinasas de proteínas implicadas en la hiperfosforilación de RyR2 (es decir, CaMKII y PKA) están sujetas a activación redox, por lo que las alteraciones en el entorno redox de los miocitos con insuficiencia cardíaca también pueden ejercer efectos indirectos sobre RyR2. En este sentido, es interesante observar que el aumento de especies reactivas de oxígeno (ROS) en la insuficiencia cardíaca también afecta negativamente a la función de SERCA2a y a otros aspectos del manejo del Ca2+ en la célula con insuficiencia cardíaca (véase la figura 2). Más recientemente, se ha demostrado que un aumento en la fuga de calcio de RyR2 produce una sobrecarga de calcio mitocondrial, lo que a su vez se ha relacionado con la progresión de la insuficiencia cardíaca.
Contribución del manejo deficiente del Ca2+ a la arritmia
La alteración en el manejo del Ca2+ en los cardiomiocitos no solo provoca disfunción sistólica y diastólica, sino que también contribuye al desarrollo de arritmias en la insuficiencia cardíaca. La desregulación de la liberación de Ca2+ mediada por RyR2 es de particular relevancia en este sentido. El aumento de la fuga diastólica de Ca2+ del retículo sarcoplásmico (RS) en las células con insuficiencia cardíaca y los consiguientes incrementos descoordinados, tanto espacial como temporalmente, del Ca2+ intracelular pueden impulsar al intercambiador Na+/Ca2+ (NCX) a intercambiar Ca2+ por Na+, induciendo así una entrada de carga positiva (Iti) que despolariza parcialmente la célula durante la fase 4 del potencial de acción (véase la figura 3). Estas despolarizaciones espontáneas del potencial de membrana se denominan posdespolarizaciones retardadas (DAD) y son arritmogénicas. Dado que la expresión y la actividad del NCX aumentan en la insuficiencia cardíaca, la entrada despolarizante de carga positiva (Iti) será mayor para cualquier liberación espontánea de Ca2+ del RS. Así, la combinación de la fuga de RyR2 y el aumento de la actividad de NCX actúa sinérgicamente para potenciar la arritmogénesis ventricular. Mecanismos similares también pueden contribuir al desarrollo de la fibrilación auricular, cuya incidencia es mayor en la insuficiencia cardíaca. Existe un potencial significativo de retroalimentación entre los diferentes mecanismos iónicos que contribuyen a la arritmogénesis en el cardiomiocito disfuncional y, por lo tanto, la posibilidad de un círculo vicioso autosostenible de alteración en el manejo de Ca2+ y Na+ que contribuye a la disfunción contráctil y la arritmia. Por ejemplo, si el NCX comienza a funcionar en modo inverso debido a una elevada concentración intracelular de [Na+], el aumento resultante en el influjo de Ca2+ puede activar quinasas proarrítmicas como CaMKII y provocar una mayor fuga de Ca2+ del retículo sarcoplásmico a través de la fosforilación de RyR2.
Disfunción sarcomérica en la insuficiencia cardíaca
La actividad mecánica cardíaca se produce como resultado de la interacción entre los cambios en la concentración de Ca2+ citosólico y los miofilamentos contráctiles (Fig. 4). La función contráctil está influenciada no solo por los cambios en la concentración de Ca2+, sino también por la respuesta intrínseca de los miofilamentos al Ca2+, la cual depende de las propiedades del complejo actomiosina y de proteínas reguladoras como el complejo troponina y la proteína C de unión a la miosina. Durante la diástole, la interacción mecánica entre la actina y la miosina está inhibida por el complejo tropomiosina-troponina. Cuando la concentración de Ca2+ citosólico aumenta durante la sístole, la unión del Ca2+ a la troponina C (cTnC) alivia este efecto inhibidor y permite que se produzca la interacción actomiosina y la contracción. Una disminución posterior del Ca2+ citosólico provoca su liberación de la cTnC y la relajación muscular. Otros componentes del sarcómero también afectan las propiedades mecánicas del músculo cardíaco. En particular, la proteína filamentosa gigante titina, que conecta el disco Z con la banda M y confiere una «elasticidad» significativa al sarcómero, influye de manera importante en la rigidez muscular pasiva, que a su vez es un determinante importante de la función diastólica.
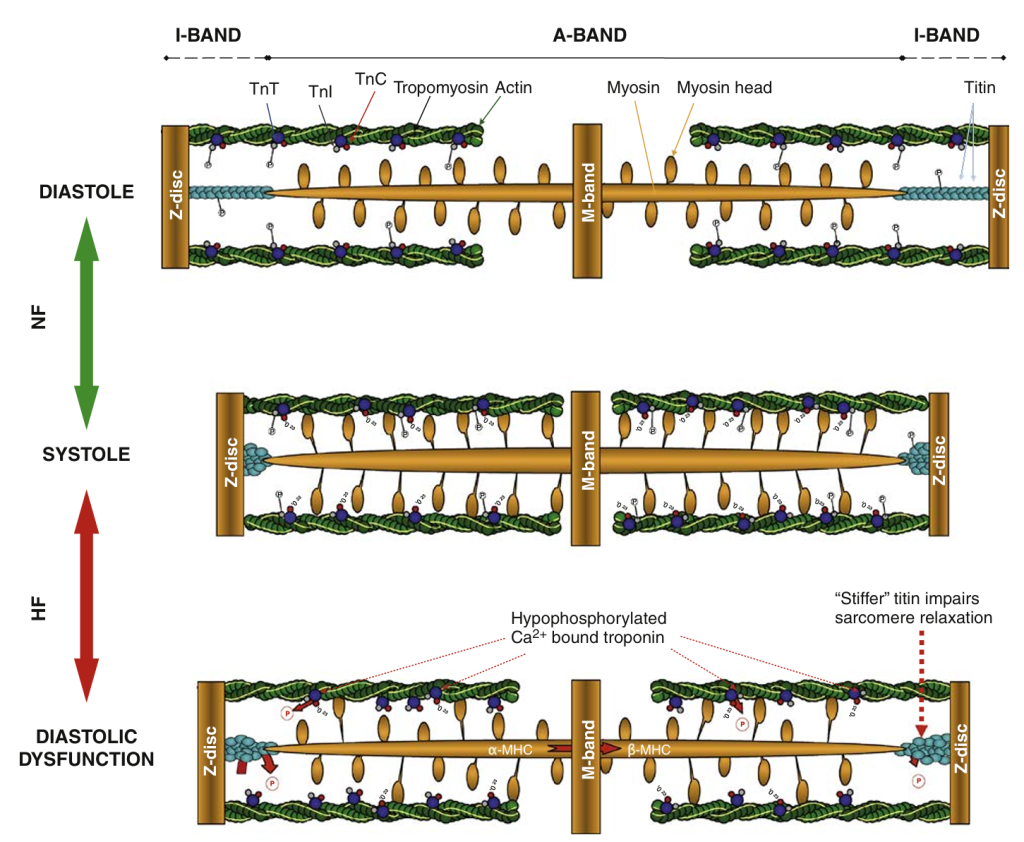
Numerosas evidencias sugieren que las alteraciones en las propiedades de los miofilamentos contribuyen a la disfunción contráctil sistólica y diastólica en la insuficiencia cardíaca. Quizás la contribución más conocida de las propiedades de los miofilamentos a los cambios contráctiles en el corazón estresado e insuficiente sea el cambio de la cadena pesada de miosina (MHC) de la isoforma rápida α-MHC a la isoforma más lenta β-MHC. La actividad ATPasa significativamente menor de la β-MHC es beneficiosa, ya que es más eficiente energéticamente, pero al mismo tiempo puede resultar en una relajación más lenta y una menor contractilidad. Este cambio de isoforma es una característica prominente en modelos de roedores, donde la α-MHC es la isoforma dominante en el miocardio sano, pero puede ser menos importante en la insuficiencia cardíaca humana, ya que la β-MHC predomina sobre la α-MHC en el miocardio ventricular humano sano. Sin embargo, se sugiere un pequeño impacto de dicho cambio de isoforma de MHC en el tejido ventricular humano con insuficiencia cardíaca.
Un regulador clave de la sensibilidad al Ca2+ de los miofilamentos en el corazón sano es la fosforilación de la troponina I (cTnI) mediada por la PKA, lo que resulta en una menor afinidad por el Ca2+ y contribuye a una cinética más rápida del ciclo de los puentes cruzados de los miofilamentos (es decir, una contracción y relajación más rápidas de los miocitos). En el corazón con insuficiencia cardíaca, la fosforilación de la cTnI dependiente de la PKA generalmente disminuye (relacionada con una menor respuesta β-adrenérgica) y resulta en una mayor sensibilidad al Ca2+ de los miofilamentos. Se cree que el principal efecto de este aumento en la sensibilidad al Ca2+ de los miofilamentos es el deterioro de la relajación y la agravación de la cinética lenta de la disminución del transitorio de Ca2+ en el miocito con insuficiencia cardíaca (véase la figura 4). Se han descrito efectos funcionales similares sobre la sensibilidad al Ca2+ de los miofilamentos tras la truncación del extremo C-terminal de la cTnI después de una lesión por isquemia/reperfusión miocárdica, y estos podrían desempeñar un papel importante en la insuficiencia cardíaca isquémica. Se cree que la fosforilación de la proteína C de unión a la miosina participa en el aumento fisiológico de la velocidad de contracción y relajación observado tras la estimulación β-adrenérgica. Por lo tanto, la hipofosforilación de la proteína C de unión a la miosina también podría contribuir a la disfunción contráctil.
Otro cambio en la isoforma proteica que se observa en corazones con insuficiencia cardíaca es la transición de la isoforma N2B de titina, más rígida (es decir, menos elástica), a la isoforma N2BA, más flexible. Se sugiere que este cambio podría contrarrestar la disminución de la fosforilación de la titina en el corazón humano con insuficiencia cardíaca, lo que funcionalmente resulta en un aumento de la rigidez pasiva. Se cree que el defecto subyacente en la fosforilación de la titina depende de la PKA (al igual que la reducción de la fosforilación de la cTnI y la proteína C de unión a la miosina), pero también se ha descrito una fosforilación disfuncional de la titina dependiente de la CaMKII. La proteína quinasa dependiente de GMP cíclico (PKG), activada por el óxido nítrico (NO), también puede fosforilar la cTnI y la titina, y tener efectos similares a los de la PKA. En la insuficiencia cardíaca, suele haber una reducción de la actividad de NO/PKG, lo que promueve un aumento de la rigidez pasiva dependiente de la titina. Finalmente, los cambios en el entorno redox dentro de los miocitos que fallan también pueden contribuir a la disfunción contráctil (por ejemplo, a través de modificaciones oxidativas específicas en la titina que conducen a una mayor rigidez pasiva).
Mecanismos globales que afectan la función de los cardiomiocitos en la insuficiencia cardíaca
Homeostasis redox en el corazón
Las especies reactivas de oxígeno (ROS) se generan en los miocitos cardíacos (al igual que en otros tipos celulares) como subproducto de la respiración y el metabolismo celular o mediante enzimas especializadas. Una función fisiológica importante de las ROS es la señalización redox (es decir, la modificación de oxidación/reducción altamente específica y generalmente reversible de moléculas de señalización implicadas en diversos procesos homeostáticos). La señalización redox está estrictamente regulada de forma espacial y temporalmente limitada, y depende de la inactivación o eliminación adecuadas de las ROS por los antioxidantes celulares para finalizar la señal. Otra función crítica de las ROS es su participación en el plegamiento oxidativo de proteínas en el retículo endoplasmático (RE). En situaciones patológicas como la insuficiencia cardíaca, las vías de señalización redox fisiológicas pueden verse alteradas o pueden activarse diferentes vías de señalización sensibles al redox. La alteración de la homeostasis redox en el RE puede tener un impacto importante en la síntesis de proteínas y las respuestas al estrés. Además, un desequilibrio entre la producción de especies reactivas de oxígeno (ROS) y las reservas antioxidantes (es decir, estrés oxidativo) puede provocar efectos perjudiciales inespecíficos debido a la oxidación irreversible de macromoléculas, membranas y ADN. Por lo tanto, las alteraciones en la homeostasis redox en el cardiomiocito disfuncional tienen un profundo impacto en muchos aspectos del fenotipo del miocito.
Las principales fuentes de especies reactivas de oxígeno (ROS) en los miocitos cardíacos con insuficiencia cardíaca incluyen la cadena de transporte de electrones mitocondrial (CTE), las proteínas NADPH oxidasa (NOX), las monoaminooxidasas, las óxido nítrico sintasas (NOS) desacopladas y las xantina oxidasas. Las especies de ROS fisiopatológicamente importantes incluyen el superóxido, el peróxido de hidrógeno (producido por la dismutación del superóxido), los iones hidroxilo y el peroxinitrito generado por la reacción del superóxido con el óxido nítrico (NO). La fuga de electrones de la CTE mitocondrial genera superóxido, y este se vuelve cuantitativamente más importante en la insuficiencia cardíaca como resultado de la disfunción de la CTE, el desacoplamiento y el deterioro de los antioxidantes mitocondriales. La generación de ROS mitocondriales puede provocar la apertura del poro de transición de permeabilidad mitocondrial (MPTP) y la pérdida de viabilidad celular. Además, dicha producción de ROS también puede inducir una mayor producción mitocondrial de ROS, denominada liberación de ROS inducida por ROS. Las monoaminooxidasas, que catabolizan los neurotransmisores noradrenalina y serotonina, se han reconocido recientemente como fuentes mitocondriales adicionales importantes de ROS en la insuficiencia cardíaca. Las fuentes no mitocondriales de ROS, como las NOX y la xantina oxidasa, también pueden estimular la producción mitocondrial de ROS. Las ROS derivadas de la xantina oxidasa pueden ser particularmente importantes en el contexto de la isquemia-reperfusión. La inhibición de las ROS mitocondriales mediante diversos agentes dirigidos a las mitocondrias se considera un enfoque terapéutico prometedor en la insuficiencia cardíaca.
Las proteínas NOX son especialmente importantes para la señalización redox, siendo la única fuente con una función primaria de generación de especies reactivas de oxígeno (ROS). Catalizan la transferencia de electrones del NADPH al O₂ molecular, generando así superóxido y/o peróxido de hidrógeno. Entre los siete miembros distintos de la familia NOX, NOX2 y NOX4 se expresan en cardiomiocitos y otras células cardíacas (p. ej., células endoteliales, fibroblastos, células inflamatorias). Aunque ambas isoformas generan ROS, existen diferencias significativas en su estructura, activación y localización subcelular que contribuyen a importantes diferencias en su función. Se ha demostrado que NOX2 tiene una función fisiológica en el acoplamiento excitación-contracción inducido por estiramiento,⁴⁰ mientras que NOX4 puede regular la diferenciación de los cardiomiocitos.⁴¹ La actividad de NOX2 y NOX4 aumenta en la insuficiencia cardíaca; la primera, principalmente como resultado de una mayor activación por estímulos como la angiotensina II y las citoquinas, y la segunda, en gran medida, debido a un aumento en sus niveles de expresión. Es posible que otra isoforma sensible al Ca2+, la NOX5, sea importante en el corazón humano.
Las enzimas NOS normalmente catalizan la producción de NO a partir de L-arginina. Los cardiomiocitos expresan constitutivamente isoformas neuronales (nNOS) y endoteliales (eNOS), las cuales tienen acciones fisiológicas distintas, especialmente en la regulación del acoplamiento excitación-contracción y la respuesta inotrópica. Una isoforma inducible de iNOS se sobreexpresa en respuesta a la estimulación con citocinas. Durante la insuficiencia cardíaca, las NOS pueden desacoplarse y pasar de la generación de NO a la de superóxido, lo que resulta en la pérdida de los efectos normales mediados por el NO, así como en efectos perjudiciales relacionados con la producción de especies reactivas de oxígeno (ROS). El desacoplamiento parcial produce la generación simultánea de NO y superóxido, lo que conduce a la producción de peroxinitrito. El desacoplamiento de las NOS suele estar relacionado con una reducción en la disponibilidad del cofactor tetrahidrobiopterina. En el caso de la eNOS, además se produce la S-glutationilación de residuos de cisteína específicos en el dominio reductasa. Es importante destacar que ambos mecanismos se ven potenciados por el estrés oxidativo, de modo que el desacoplamiento de la NOS puede, a menudo, amplificar la generación de especies reactivas de oxígeno (ROS) por otras fuentes.
Las alteraciones en el equilibrio antioxidante contribuyen significativamente a la alteración de la homeostasis redox en el corazón con insuficiencia cardíaca. Estas alteraciones incluyen cambios en enzimas antioxidantes como la superóxido dismutasa, la catalasa, la glutatión peroxidasa, la tiorredoxina, la peroxirredoxina, las glutatión S-transferasas y otras. Un factor crucial para la homeostasis redox es el nivel de NADPH en los diferentes compartimentos celulares, ya que este nucleótido es necesario para la regeneración de las reservas reducidas de los principales antioxidantes celulares, como el glutatión, la glutaredoxina y la tiorredoxina. Por lo tanto, las reacciones metabólicas que generan NADPH tienen un impacto potencialmente amplio en el fenotipo del miocito. En las mitocondrias, la actividad de las deshidrogenasas dependientes de Ca2+ es importante en este sentido, y esta actividad se ve inhibida por la sobrecarga de Na+ citosólico en la insuficiencia cardíaca, como se mencionó anteriormente. En el citosol, la glucosa-6-fosfato deshidrogenasa (G6PD) es una enzima limitante clave involucrada en la producción de NADPH y se ha demostrado que afecta la homeostasis del calcio de los cardiomiocitos y la disfunción contráctil en la isquemia-reperfusión. Curiosamente, los niveles excesivamente altos de NADPH y glutatión pueden ser perjudiciales al inducir el llamado estrés reductivo. En un modelo de ratón de cardiomiopatía por αB-cristalina mutante (CryAB), dicho estrés reductivo se asocia con un plegamiento anormal de proteínas y la acumulación de agregados proteicos en los cardiomiocitos.
La alteración de la homeostasis redox afecta a una amplia gama de dianas moleculares en el cardiomiocito disfuncional, incluyendo quinasas, fosfatasas, transportadores y canales iónicos, miofilamentos y factores de transcripción, como se revisa en detalle en otras publicaciones. Aquí analizamos algunos ejemplos donde los mecanismos de dichas modificaciones redox y su impacto en el fenotipo del miocito se han abordado con suficiente profundidad como para ofrecer la posibilidad de identificar dianas terapéuticas viables. La desregulación redox afecta a varias proteínas implicadas en el acoplamiento excitación-contracción anómalo, como se mencionó anteriormente. La NOX2 parece ser una fuente importante de especies reactivas de oxígeno (ROS) en este sentido y puede actuar tanto mediante la oxidación directa de proteínas cruciales en el acoplamiento excitación-contracción, como RyR2, como a través de la modulación de quinasas o fosfatasas sensibles al redox que regulan dichas proteínas. La NOX2 también está implicada en la génesis de la fibrilación auricular. Varias vías de señalización implicadas en la hipertrofia de los cardiomiocitos están parcialmente reguladas por el estado redox (p. ej., la activación de ASK-1, ERK1/2, NF-κB y Akt). NOX2 es, de nuevo, una importante fuente de ROS responsable de dicha regulación, especialmente en el contexto de una mayor activación del sistema renina-angiotensina. Las histonas deacetilasas de clase II (HDAC) son reguladores clave de la expresión génica hipertrófica, permitiendo que esto ocurra cuando se exportan del núcleo del cardiomiocito. Se ha descrito una vía sensible a la tiorredoxina 1 para la oxidación específica y posterior exportación nuclear de HDAC4 como importante en la hipertrofia de los cardiomiocitos mediada por la vía adrenérgica. La viabilidad de los cardiomiocitos puede verse afectada negativamente por un desequilibrio redox (p. ej., lo que conduce a una mayor producción mitocondrial de ROS). Además, se ha descubierto que la activación oxidativa específica de quinasas como CaMKII es importante. La PKG también se activa oxidativamente y se ha demostrado que es fisiológicamente importante en la modulación de la respuesta de Frank-Starling inducida por estiramiento mediante la fosforilación de la fosfolambana, pero se ha observado que la oxidación persistente durante la sobrecarga de presión crónica es perjudicial. No todos los efectos de las especies reactivas de oxígeno (ROS) son perjudiciales en el cardiomiocito sometido a estrés. Por ejemplo, se ha demostrado que un aumento de NOX4 es cardioprotector durante la sobrecarga de presión crónica o el estrés isquémico, al potenciar mecanismos de señalización beneficiosos, incluida la señalización del factor inducible por hipoxia 1-alfa (HIF1α) en el miocito, lo que conduce a efectos paracrinos sobre la densidad capilar miocárdica, potenciación de la respuesta integrada al estrés mediada por el factor de transcripción activador 4 (ATF4) y alteraciones en el uso de sustratos miocárdicos. Esto subraya el concepto de que los efectos de la alteración de la homeostasis redox pueden variar según la fuente de ROS y su localización subcelular.
Síntesis, recambio, control de calidad y respuestas al estrés de las proteínas.
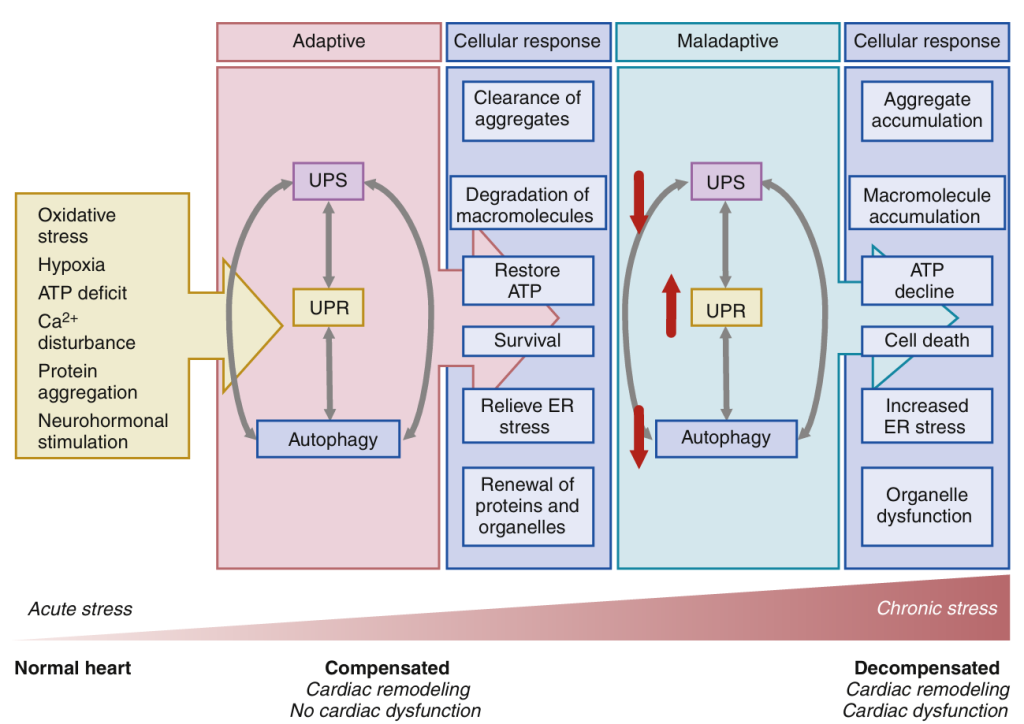
La remodelación celular dinámica que ocurre en el corazón estresado e insuficiente implica cambios sustanciales en la síntesis y el recambio de proteínas, y requiere mecanismos robustos de control de calidad para asegurar la degradación o eliminación de proteínas anormales (p. ej., proteínas mal plegadas y agregados proteicos). Varios orgánulos y sistemas son importantes para estas funciones, incluyendo el RE, el sistema ubiquitina-proteasoma (UPS) y la autofagia. La célula puede generar respuestas específicas al estrés diseñadas para facilitar el mantenimiento de algunas de estas funciones (p. ej., la respuesta a proteínas mal plegadas [UPR] o una mejora de la autofagia). Estos sistemas contribuyen significativamente al fenotipo celular en el corazón insuficiente y pueden representar dianas para la modulación terapéutica. A continuación, analizaremos con mayor detalle el papel de algunos de estos sistemas en el corazón insuficiente.
Retículo endoplasmático y respuesta a proteínas mal plegadas
El retículo sarcoplásmico (RS) del miocito cardíaco es un retículo endoplasmático (RE) especializado, especialmente importante para la regulación intracelular del Ca2+, y se sabe que su función se ve afectada en la insuficiencia cardíaca, como se mencionó anteriormente en este capítulo. Además de la homeostasis del Ca2+, el RE coordina otras funciones importantes, como el correcto plegamiento, la modificación postraduccional y el control de calidad de las proteínas de membrana y secretadas, la gluconeogénesis, la síntesis de lípidos y esteroides. También interactúa con la función mitocondrial e influye en ella (por ejemplo, mediante la transferencia de Ca2+ desde el RE). Estas funciones son importantes en los miocitos cardíacos, al igual que en otros tipos celulares, aunque la distinción o superposición entre el RS y el RE aún no está bien definida. La necesidad de plegamiento proteico, control de calidad y funciones relacionadas en el RE cambia significativamente en el corazón remodelado, donde se producen alteraciones importantes en el recambio proteico (por ejemplo, debido a la hipertrofia), el metabolismo y la energética. Por lo tanto, el mantenimiento de una función adecuada del RE se convierte en un factor importante en la homeostasis celular.
La respuesta al estrés del retículo endoplasmático (RPE) es un mecanismo conservado que detecta el estado del plegamiento de las proteínas del retículo endoplasmático al identificar la acumulación de proteínas mal plegadas e inducir un programa de cambios que afecta tanto al retículo endoplasmático como a otros orgánulos y procesos celulares. La RPE abarca mecanismos homeostáticos que incluyen el aumento de la síntesis de chaperonas, las cuales ayudan al correcto plegamiento de las proteínas mal plegadas; la inhibición de la traducción de proteínas que podría agravar el mal plegamiento y la sobrecarga del retículo endoplasmático; y la activación de vías de degradación para descomponer las proteínas mal plegadas. Diversos factores estresantes pueden inducir la RPE, como la privación de glucosa o aminoácidos, el estrés oxidativo, la hipoxia y el desequilibrio iónico. La inducción de la UPR está mediada por tres sensores transmembrana de estrés del RE: la proteína quinasa similar al ARN del RE (PERK), la quinasa 1 dependiente de inositol (IRE1) y el factor de transcripción activador 6 (ATF6). Estos sensores detectan el estrés del RE luminal y responden activando vías transcripcionales específicas e inhibiendo la traducción global de proteínas (Fig. 5). Dicha activación de la UPR puede ser una respuesta adaptativa que restablece la homeostasis o una respuesta perjudicial que desencadena vías de muerte celular, dependiendo de su intensidad, duración y contexto.
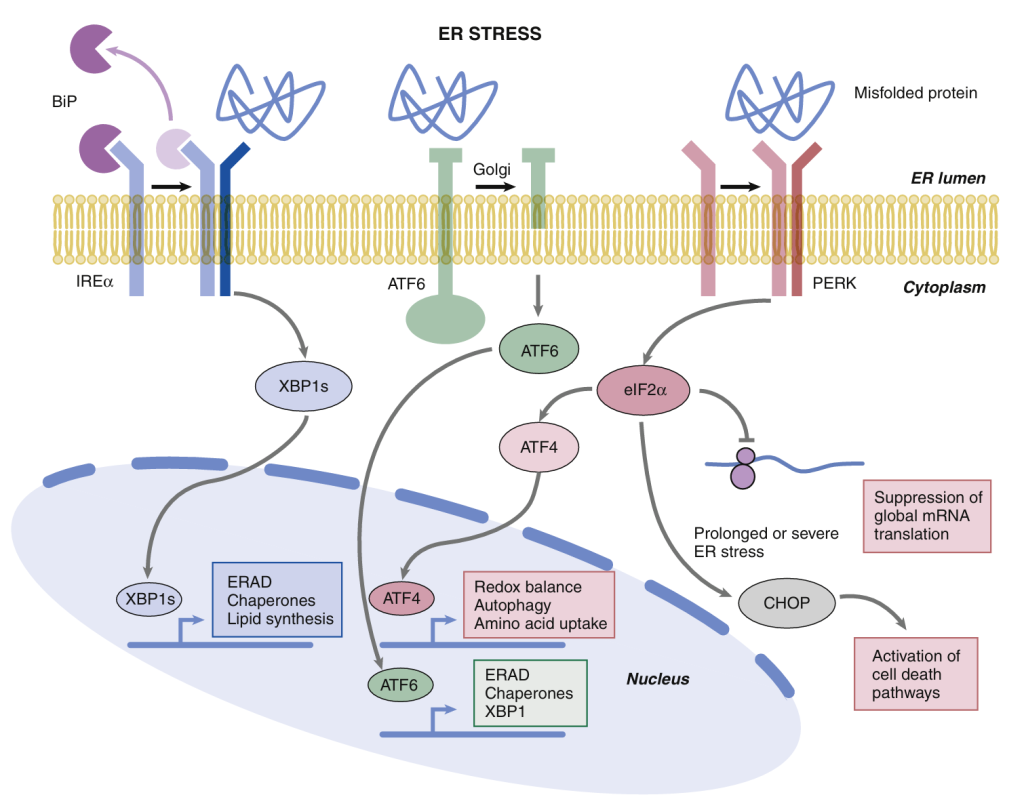
Investigaciones recientes indican que la UPR se activa en el corazón isquémico, hipertrófico e insuficiente, y contribuye al fenotipo celular a través de sus efectos sobre el recambio proteico, el metabolismo, la función y la viabilidad celular. El tejido miocárdico de pacientes con miocardiopatía dilatada o isquémica muestra una mayor expresión de muchas proteínas implicadas en la UPR, como los tres sensores de estrés y las chaperonas del RE. Se observa una activación similar de la UPR en modelos animales de insuficiencia cardíaca (por ejemplo, ratones sometidos a sobrecarga de presión crónica o en modelos de hipertensión e insuficiencia cardíaca sensibles a la sal). La expresión transgénica de un receptor KDEL mutante, cuya función normal es recuperar chaperonas del RE y proteínas mal plegadas con secuencias KDEL (LysAsp-Glu-Leu) de compartimentos intracelulares, como el aparato de Golgi, hasta el RE, como parte de un mecanismo de control de calidad, produce cardiomiopatía dilatada en un modelo de ratón, lo que indica la importancia de la función de las chaperonas del RE.⁶¹ Evidencia más directa del papel de la UPR en la insuficiencia cardíaca proviene de estudios en ratones en los que la expresión específica en el corazón de un mutante dominante negativo de ATF6 resultó en remodelación cardíaca adversa e insuficiencia cardíaca, mientras que los ratones que expresaban un ATF6 constitutivamente activo presentaban una remodelación cardíaca reducida tras un infarto de miocardio. Se observó que la activación de un programa genético dependiente de ATF6 por la trombospondina 4 era protectora durante la lesión miocárdica. ATF6 también parece proteger contra el estrés cardíaco agudo, como la lesión por isquemia-reperfusión. Estos estudios sugieren que la activación de la vía ATF6 de la UPR podría ser una respuesta adaptativa durante la insuficiencia cardíaca. Como se mencionó anteriormente, la activación de ATF4 mediada por NOX4 también puede ser beneficiosa. Sin embargo, también puede haber situaciones en las que la activación de UPR sea perjudicial.
Los datos previos sugieren que modular la UPR, o componentes específicos de la misma, podría ser un enfoque terapéutico beneficioso para abordar la disfunción celular en la insuficiencia cardíaca. Por ejemplo, se podrían diseñar estrategias para potenciar la activación de ATF6. Otra alternativa sería utilizar chaperonas químicas artificiales como el ácido 4-fenilbutírico (4-PBA) y el ácido tauroursodesoxicólico (TUDCA). Se ha demostrado que estas sustancias reducen el estrés del retículo endoplasmático y tienen efectos beneficiosos en modelos de resistencia a la insulina. Estudios preliminares también sugieren efectos beneficiosos en modelos murinos de sobrecarga de presión crónica.
Autofagia
La autofagia (autodigestión) es un importante mecanismo homeostático para la degradación y el reciclaje de diversos componentes celulares, incluyendo orgánulos (p. ej., mitocondrias, RE), macromoléculas (p. ej., agregados proteicos) y lípidos. Cumple una importante función de control de calidad, además de ser un mecanismo que genera nuevos componentes básicos para procesos celulares como la producción de energía y la síntesis de proteínas, nucleótidos y membranas (p. ej., durante la inanición). Se describen diferentes formas de autofagia, entre las cuales la macroautofagia (aquí denominada simplemente autofagia) es la más frecuente y mejor estudiada. El proceso de autofagia está altamente regulado y se describe en detalle en otras publicaciones. Se caracteriza por la formación de autofagosomas de doble membrana alrededor de orgánulos dañados o agregados proteicos, que luego se fusionan con lisosomas para formar autofagolisosomas, dentro de los cuales el contenido es degradado por hidrolasas lisosomales. Una familia de proteínas codificadas por genes relacionados con la autofagia (Atg) participa en las diferentes etapas del proceso autofágico. El complejo 1 de la diana de rapamicina en mamíferos (mTORC1) es un activador clave de la autofagia en respuesta al «estrés nutricional» (por ejemplo, hipoxia o privación de nutrientes).
Se han encontrado niveles elevados de autofagia en corazones humanos con insuficiencia cardíaca, y estudios en modelos de ratones genéticamente modificados han proporcionado información mecanicista sobre las funciones fisiológicas y patológicas de la autofagia en el corazón. Los ratones que carecen de la proteína de membrana asociada al lisosoma 2 (LAMP2), necesaria para la degradación autofágica, desarrollan cardiomiopatía. De manera similar, los ratones con deficiencia cardíaca específica de Atg5 desarrollan disfunción cardíaca acompañada de niveles elevados de proteínas ubiquitinadas, estrés del retículo endoplasmático y apoptosis, y mueren prematuramente después de aproximadamente 6 meses de edad. Además, estos animales presentan una respuesta deficiente a la sobrecarga de presión, lo que sugiere que la autofagia es un mecanismo de protección adaptativo. Sin embargo, puede haber algunas situaciones en las que la autofagia sea perjudicial.
Sistema ubiquitina-proteasoma
El sistema ubiquitina-proteasoma (UPS) es el principal sistema celular para la degradación de proteínas, tanto como vía fisiológica (p. ej., en diversas respuestas de señalización celular) como en situaciones donde hay un aumento de proteínas mal plegadas o dañadas (p. ej., estrés del retículo endoplasmático o estrés oxidativo). Al igual que la autofagia, la degradación de proteínas dependiente del UPS es un proceso altamente regulado. Las proteínas son dirigidas a la degradación dependiente de ATP en el proteasoma 26S después de haber sido poliubiquitinadas en residuos de lisina mediante enzimas específicas activadoras de ubiquitina, conjugadoras de ubiquitina y ubiquitina ligasas.
Se ha observado que las proteínas ubiquitinadas se acumulan en el miocardio humano con insuficiencia cardíaca, y que la actividad catalítica del proteasoma se reduce notablemente en comparación con corazones sanos. Sin embargo, el impacto funcional no queda claro en estos estudios. Los estudios en modelos animales de insuficiencia cardíaca presentan resultados contradictorios: algunos muestran una disminución y otros un aumento de la actividad del sistema ubiquitina-proteasoma (UPS). De interés clínico, pequeños estudios en pacientes con dispositivos de asistencia ventricular izquierda (DAVI) han reportado que la actividad del proteasoma aumenta en asociación con la remodelación inversa inducida por la descarga mecánica, lo que sugiere que el UPS podría tener un papel beneficioso, quizás eliminando proteínas dañadas. La evidencia que respalda la importancia de la eliminación de proteínas dañadas en el corazón con insuficiencia cardíaca proviene de modelos de miocardiopatía relacionada con la desmina y de mutaciones en la chaperona αB-cristalina (CryAB), donde la acumulación de agregados proteicos parece ser proteotóxica para el corazón. La miocardiopatía relacionada con la desmina humana puede reproducirse en ratones que expresan una mutación CryABR120G específica del corazón; estos animales muestran evidencia de agregación de proteínas, alteración del sistema ubiquitina-proteasoma y miocardiopatía dilatada.
Funciones coordinadas de la respuesta a proteínas mal plegadas, el sistema ubiquitina-proteasoma y los sistemas de autofagia.
Los sistemas UPR, UPS y autofagia (y otros sistemas de degradación de proteínas, como la vía de la calpaína, que no se abordan aquí) actúan de forma coordinada para regular el recambio proteico. Los tres sistemas se activan por estímulos similares asociados al estrés cardíaco y la insuficiencia cardíaca (p. ej., estrés oxidativo, hipoxia, cambios metabólicos y desregulación del Ca2+). Las alteraciones en la función de un sistema suelen afectar a los demás. Por ejemplo, el estrés del RE puede activar el UPS y la autofagia, mientras que la activación del UPS puede aumentar cuando la autofagia está alterada. Además, estos sistemas tienen un impacto más amplio en otros procesos, como el metabolismo energético, el estado redox y la homeostasis iónica, y por lo tanto pueden afectar a muchos aspectos diferentes del fenotipo celular en el corazón con insuficiencia cardíaca. Como se mencionó anteriormente, el funcionamiento adecuado de estas vías parece ser necesario para la adaptación al estrés cardíaco crónico y el mantenimiento de la función cardíaca normal. Por otro lado, una activación insuficiente o excesiva de estas vías puede contribuir a la remodelación desadaptativa y al desarrollo de insuficiencia cardíaca (Fig. 6).
Interacciones de los cardiomiocitos con otros tipos de células
El número de células no miocárdicas (p. ej., fibroblastos, células endoteliales) en el corazón supera al de las células contráctiles, y las interacciones entre cardiomiocitos y células no miocárdicas son importantes no solo fisiológicamente, sino también en el contexto de la insuficiencia cardíaca. En este trabajo nos centramos en la señalización paracrina intercelular que involucra a los cardiomiocitos durante el desarrollo de la remodelación y la insuficiencia cardíaca.
Efectos paracrinos de las células endoteliales
Se sabe que las células endoteliales microvasculares coronarias y endocárdicas ejercen efectos paracrinos sobre los cardiomiocitos mediante la secreción de factores como el NO, la endotelina y la neuregulina, afectando tanto la función contráctil como el crecimiento. Por ejemplo, el NO modula la relajación miocárdica, la respuesta de Frank-Starling, la respuesta inotrópica β-adrenérgica y la energética miocárdica. Los efectos dependientes del NO endotelial sobre la función miocárdica pueden verse afectados durante la sobrecarga de presión crónica y la insuficiencia cardíaca (por ejemplo, debido a un aumento en la producción de especies reactivas de oxígeno endoteliales), lo que repercute en la función contráctil, el metabolismo de los sustratos y la hipertrofia. La producción endotelial de endotelina-1 (ET-1) actúa como estímulo para la hipertrofia de los cardiomiocitos durante la sobrecarga de presión.
Efectos paracrinos de los fibroblastos
Los fibroblastos cardíacos desempeñan un papel fundamental no solo en la fibrosis, sino también en la regulación de la hipertrofia de los cardiomiocitos. Su fenotipo cambia en respuesta al estrés y se transforman en miofibroblastos que secretan factores paracrinos cardioactivos. La evidencia experimental sugiere que un componente significativo de los efectos hipertróficos cardíacos mediados por la angiotensina II puede estar mediado por factores de fibroblastos, como la endotelina-1 (ET-1) y el factor de crecimiento transformante β (TGF-β). Otros factores de crecimiento importantes en la interacción paracrina y autocrina entre cardiomiocitos y miofibroblastos incluyen el factor de crecimiento de fibroblastos 2 (FGF-2), el factor de crecimiento derivado de plaquetas (PDGF)-A/B, el factor de crecimiento similar a la insulina 1 (IGF-1) y la familia de interleucinas IL-6, que incluye el factor inhibidor de la leucemia (LIF) y la cardiotrofina-1 (CT-1). Algunas de estas sustancias son cardioprotectoras, pero muchas actúan estimulando la fibrosis en la respuesta primaria de «curación» ante una lesión.
Señalización angiogénica de los cardiomiocitos
Existe una relación importante entre la hipertrofia de los cardiomiocitos y la densidad capilar miocárdica y la angiogénesis, especialmente durante la sobrecarga de presión crónica. Trabajos experimentales recientes indican que una correspondencia entre la densidad capilar y la hipertrofia ayuda a preservar la función contráctil cardíaca y a reducir el remodelado adverso. Los estímulos hipertróficos inducen la expresión en los cardiomiocitos de factores de crecimiento angiogénicos como el VEGF, la angiopoyetina-2 y el factor de crecimiento derivado de plaquetas (PDGF), que promueven la angiogénesis y actúan para mantener el flujo sanguíneo miocárdico. La producción de VEGF por los cardiomiocitos está regulada tanto por vías de señalización hipóxicas como no hipóxicas a través de los factores de transcripción HIF1 y GATA4, respectivamente. Se ha descubierto que una inhibición de la activación de HIF1 en los cardiomiocitos durante la sobrecarga de presión crónica promueve la descompensación y el desarrollo de insuficiencia cardíaca, mientras que el VEGF exógeno es protector. Recientemente, se identificó una mejora dependiente de NOX4 de la activación de HIF1 en los cardiomiocitos y la posterior liberación de VEGF como un importante mecanismo cardioprotector endógeno durante la sobrecarga de presión crónica.
Activación de las vías inflamatorias
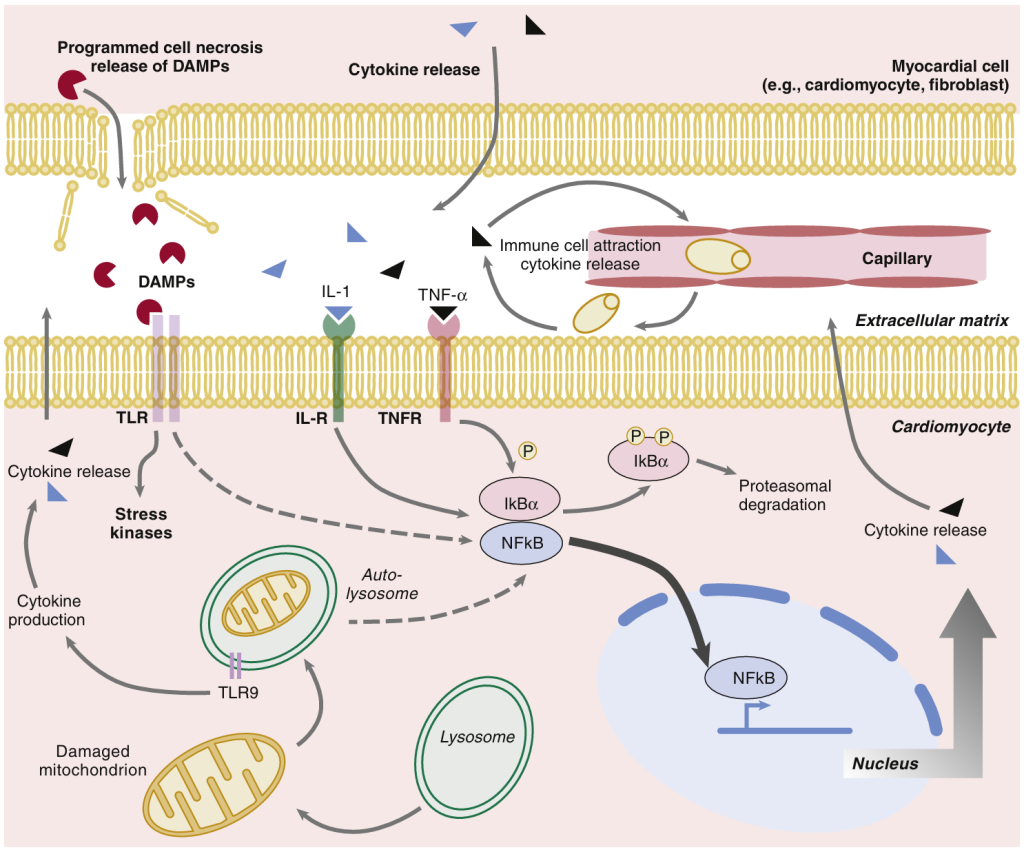
La activación del sistema inmunitario innato desempeña un papel importante en la progresión hacia la insuficiencia cardíaca. Desde hace muchos años se sabe que las citocinas proinflamatorias y antiinflamatorias producidas en el propio corazón bajo condiciones de estrés hemodinámico participan en la infiltración de células inflamatorias en el miocardio, lo que contribuye a la disfunción contráctil y al daño de los cardiomiocitos, así como a la remodelación de la matriz extracelular. El factor de necrosis tumoral (TNF), una citocina proinflamatoria, ha sido ampliamente estudiado en la progresión de la insuficiencia cardíaca. Los modelos murinos de sobreexpresión de TNF específica del corazón desarrollan hipertrofia ventricular acelerada, fibrosis, dilatación e insuficiencia cardíaca durante la sobrecarga de presión crónica. Tanto el TNF como la IL-1 activan una remodelación perjudicial dependiente de NF-κB en el corazón. A pesar de la caracterización de estas vías, las terapias dirigidas a citocinas como el TNF-α no han dado lugar a tratamientos eficaces hasta el momento.
Estudios recientes indican que un mecanismo importante para el desarrollo de la inflamación “estéril” (es decir, no infecciosa) durante la insuficiencia cardíaca podrían ser los patrones moleculares asociados al daño (DAMPs) liberados por las células miocárdicas dañadas (Fig. 7). Los DAMPs consisten en proteínas intracelulares liberadas, como las proteínas de choque térmico, las LDL oxidadas y los ácidos grasos libres, o componentes de la matriz extracelular, como la fibronectina.⁹³ Son reconocidos por las células inmunitarias a través de receptores de superficie celular, incluidos los receptores tipo Toll (TLR) y los receptores tipo NOD (dominio de oligomerización de nucleótidos), lo que desencadena respuestas inflamatorias. Los TLR también pueden activarse en los propios cardiomiocitos para inducir la producción de citocinas, que amplifican la respuesta inflamatoria. De hecho, se ha informado que el TLR4 está sobreexpresado en el corazón humano con insuficiencia cardíaca. Recientemente, se ha descrito un nuevo mecanismo que vincula la disfunción mitocondrial, la autofagia y la inflamación estéril en el corazón sometido a sobrecarga de presión crónica. Oka y colaboradores descubrieron que, durante la mitofagia (autofagia de mitocondrias disfuncionales) en corazones sometidos a sobrecarga de presión, se liberan fragmentos de ADN mitocondrial que estimulan la activación de los receptores TLR9 de los cardiomiocitos y provocan inflamación. La inactivación genética de la ADNasa II de los cardiomiocitos, enzima que degrada el ADN mitocondrial, resultó en una marcada acumulación de ADN mitocondrial en los autolisosomas, así como en una miocarditis profunda y una miocardiopatía dilatada. Es importante destacar que el tratamiento con un antagonista de TLR9 redujo la inflamación miocárdica tanto en ratones con deficiencia de ADNasa II como en ratones de tipo silvestre, lo que sugiere que este podría ser un objetivo terapéutico adecuado para la insuficiencia cardíaca.
Vías dependientes de microarn y arn no codificable largo
Los microARN (miRNA) son segmentos de nucleótidos endógenos altamente conservados de ARN no codificante (generalmente de unos 22 nucleótidos de longitud) que regulan la estabilidad y la traducción de los ARNm. Los miRNA funcionan mediante el apareamiento de bases con las regiones 3′ no traducidas de un transcrito, lo que resulta en el silenciamiento génico a través de la represión traslacional y/o la degradación del ARNm diana. Los miRNA individuales pueden actuar como «reguladores maestros» de los procesos de señalización celular gracias a su capacidad para regular múltiples genes dentro de las vías de señalización. Por lo tanto, pueden tener un impacto importante en los fenotipos celulares. Trabajos recientes en modelos animales, respaldados por estudios en tejido humano, indican que las alteraciones en la expresión de miRNA influyen significativamente en el fenotipo de los cardiomiocitos en el corazón con insuficiencia cardíaca (Fig. 8). Además, estos efectos se extienden a las células no miocárdicas y a las interacciones célula-célula dentro del corazón.
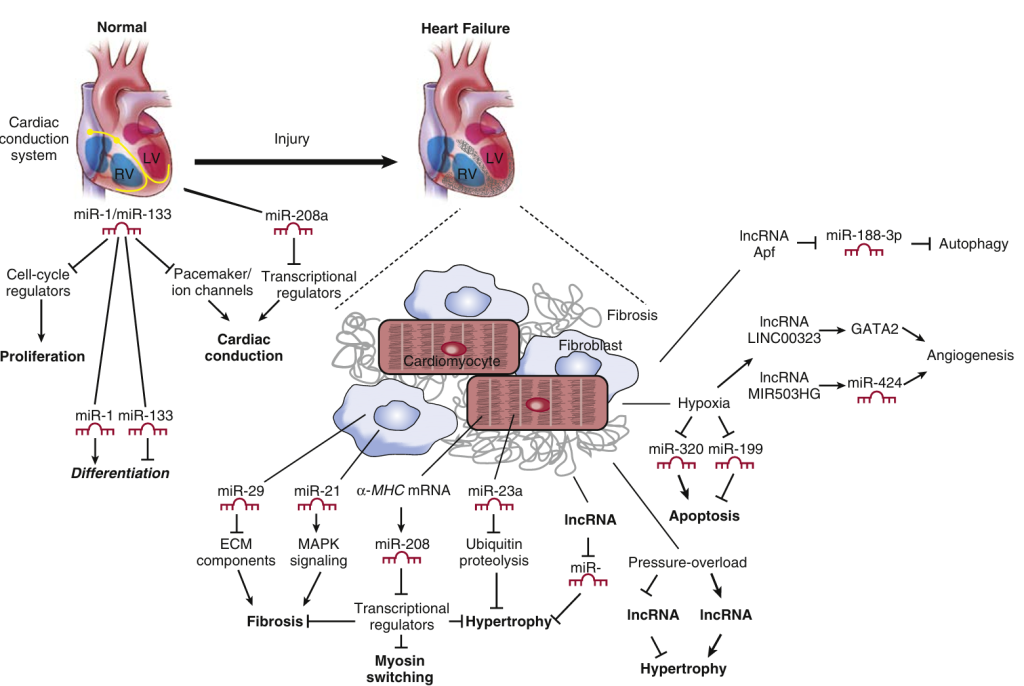
Numerosos microARN pueden influir en la respuesta hipertrófica de los cardiomiocitos al estrés, incluyendo efectos antihipertróficos (p. ej., miR-1, miR-9, miR-26, miR-29, miR-98, miR-133) y prohipertróficos (p. ej., miR-21, miR-23a, miR-143, miR-199a, miR-199b, miR-208, miR-499). Algunos de estos actúan principalmente en células no miocárdicas en el corazón en remodelación y con insuficiencia cardíaca (p. ej., miR-21 y miR-29 en fibroblastos). Los cambios en los perfiles de microARN desempeñan un papel importante en la reexpresión de los perfiles genéticos fetales en el corazón estresado y con insuficiencia cardíaca. Curiosamente, la transcripción de miRNA puede estar regulada por ciertos factores de transcripción activados por estrés (p. ej., NFAT, STAT3, SRF), lo que sugiere que los circuitos reguladores globales coordinan la remodelación molecular que sustenta el desarrollo del fenotipo celular en el corazón con insuficiencia cardíaca. Los objetivos moleculares específicos de los miRNA que se alteran en la insuficiencia cardíaca incluyen proteínas implicadas en el crecimiento de los cardiomiocitos, la función del sarcómero, el acoplamiento excitación-contracción, la regulación del ciclo celular, el recambio proteico, la homeostasis redox, la regulación paracrina, la fibrosis, la inflamación, la angiogénesis y otros procesos, lo que ilustra los efectos potencialmente profundos de estos reguladores maestros.
En principio, el aumento de la expresión de miRNA puede ser objeto de terapias (por ejemplo, con antagomirs), y estudios en modelos animales sugieren que estos enfoques ofrecen un potencial significativo. El concepto de atacar múltiples componentes patológicos con un solo antagomir resulta intuitivamente atractivo, pero existen importantes desafíos que superar, en particular la necesidad de dirigir la acción específicamente al corazón. En principio, los miRNA también podrían administrarse como terapia, pero nuevamente existen desafíos con respecto a la farmacocinética, la farmacodinámica y la especificidad de la acción.
Los microARN (miRNA) también se encuentran circulando en la sangre de forma estable, tanto en personas sanas como en personas con enfermedades. Los perfiles de miRNA en plasma cambian de forma específica y significativa en diferentes enfermedades, incluida la insuficiencia cardíaca. La relevancia fisiopatológica de este hallazgo aún no se ha establecido por completo (por ejemplo, ¿facilitan los miRNA circulantes la señalización entre diferentes células o tejidos, o simplemente representan un efecto de «desbordamiento»?). Independientemente de la respuesta, existe un gran interés en el potencial de los cambios en los perfiles de miRNA en plasma para servir como biomarcadores para el diagnóstico, el pronóstico y/o el seguimiento de enfermedades.
De forma similar, otra clase de ARN no codificantes altamente abundantes, denominados ARN largos no codificantes (lncRNA), están emergiendo como potenciales reguladores de la remodelación en el corazón con insuficiencia cardíaca. Estos suelen tener más de 200 nucleótidos de longitud y pueden desempeñar diversas funciones reguladoras en la expresión y traducción génica (por ejemplo, modulación epigenética, actuando como esponjas moleculares y modulando la señal del empalme alternativo de genes). Se ha demostrado que los lncRNA tienen funciones importantes en el desarrollo cardíaco, pero la investigación de su papel en las respuestas cardíacas al estrés y en la insuficiencia cardíaca aún se encuentra en sus inicios. No obstante, los primeros estudios sugieren que los lncRNA individuales pueden tener funciones importantes en la regulación epigenética durante la hipertrofia cardíaca, y los estudios de asociación del genoma completo en humanos también respaldan un papel clave.