Introducción
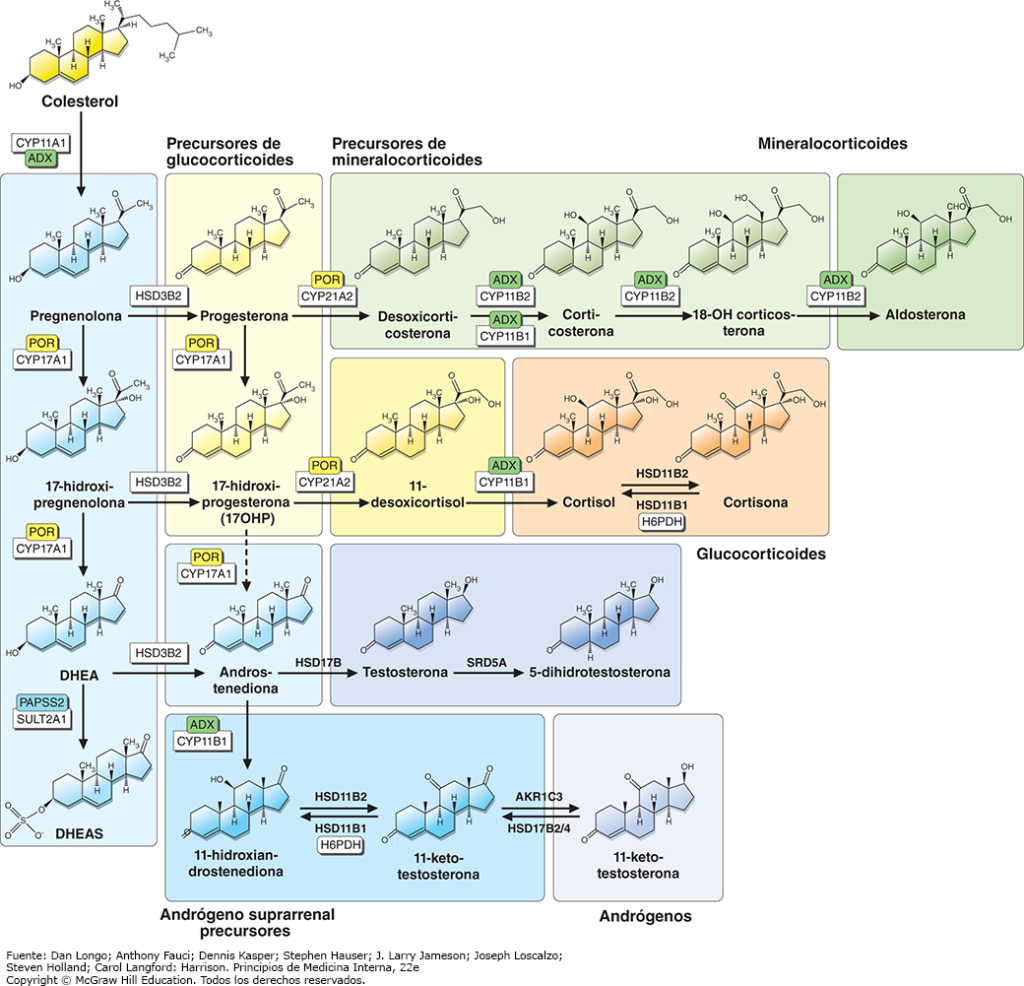
La corteza suprarrenal produce tres clases de hormonas corticoesteroides: glucocorticoides (p. ej., cortisol), mineralocorticoides (p. ej., aldosterona) y precursores de andrógenos suprarrenales (p. ej., dehidroepiandrosterona, [DHEA]) (fig. 1). Los glucocorticoides y los mineralocorticoides actúan a través de receptores nucleares específicos para regular eventos de respuesta a la tensión fisiológica, así como la presión arterial y la homeostasis electrolítica. Los precursores de los andrógenos suprarrenales se transforman en las gónadas y en las células efectoras (blanco) periféricas en esteroides sexuales, que actúan por medio de los receptores nucleares de los andrógenos y los estrógenos.
Las enfermedades de la corteza suprarrenal se caracterizan por deficiencia o exceso de una o más de las tres principales clases de corticoesteroides. La deficiencia hormonal puede ser resultado de trastornos hereditarios glandulares o enzimáticos o de la destrucción de las glándulas hipofisaria o suprarrenales por enfermedades autoinmunitarias, infecciones, infartos o episodios yatrogénicos (como cirugías o supresión hormonal). En general, el exceso de hormonas es el resultado de neoplasias, que incrementan la producción de hormona adrenocorticotrópica (ACTH, adrenocorticotropic hormone) por parte de células hipofisarias o neuroendocrinas ectópicas productoras de ACTH, o bien, por la mayor producción de glucocorticoides, mineralocorticoides o precursores de andrógenos suprarrenales por los nódulos suprarrenales o hiperplasia. Los nódulos suprarrenales se identifican cada vez con mayor frecuencia de manera incidental durante la realización de estudios capaces de generar imágenes de cortes transversales del tórax o el abdomen, realizados por otras causas.
Anatomía y desarrollo de las glándulas suprarrenales
Las glándulas suprarrenales normales pesan de seis a 11 g cada una. Se localizan encima de los riñones y tienen su propio suministro sanguíneo. La sangre arterial fluye primero a la región subcapsular y pasa de la zona glomerulosa cortical externa, atravesando la zona fasciculada intermedia, a la zona reticular interna y, por último, a la médula suprarrenal. La vena suprarrenal derecha drena de manera directa en la vena cava, en tanto que la vena suprarrenal izquierda lo hace en la vena renal izquierda.
Durante el desarrollo embrionario temprano, las glándulas suprarrenales surgen de la cresta urogenital y luego se separan de las gónadas y de los riñones, aproximadamente en la sexta semana de gestación. Al mismo tiempo que ocurre la diferenciación sexual (de la séptima a la novena semanas de gestación, la corteza suprarrenal comienza a producir cortisol y DHEA, el precursor de esteroides sexuales suprarrenales. Los receptores nucleares huérfanos del factor esteroidogénico 1 (SF1, steroidogenic factor 1, codificado por el gen NR5A1) y de la proteína del “gen de inversión sexual sensible a la dosis 1” (DAX1, dosage-sensitive sex reversal gene 1, codificada por el gen NR0B1), entre otros, son fundamentales durante este periodo del desarrollo, porque regulan muchos genes de las glándulas suprarrenales que participan en la esteroidogénesis.
Regulación de la esteroidogénesis
La producción de glucocorticoides y andrógenos suprarrenales la controla el eje hipotálamo-hipófisis-glándulas suprarrenales (HPA, hypothalamic-pituitary-adrenal), en tanto que los mineralocorticoides están regulados por el sistema renina-angiotensina-aldosterona (RAA, renin-angiotensin-aldosterone).
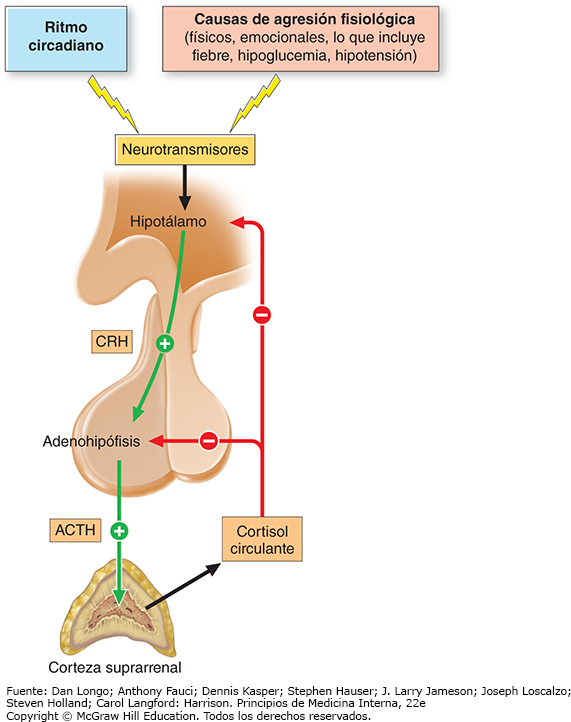
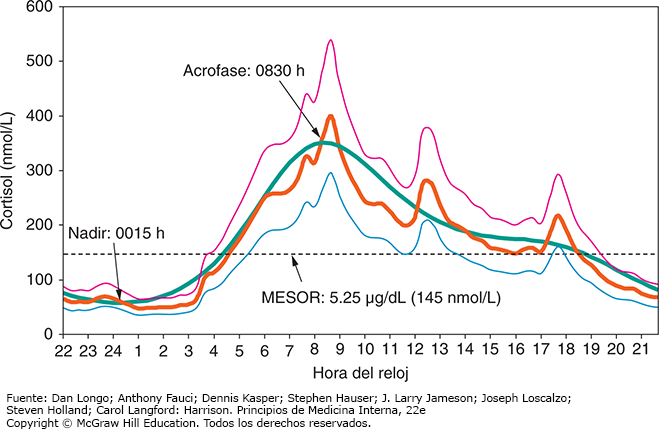
El hipotálamo y la hipófisis controlan la síntesis de glucocorticoides mediante retroalimentación inhibidora (fig. 2). La liberación hipotalámica de hormona liberadora de corticotropina (CRH, corticotropin-releasing hormone) ocurre en respuesta a estrés exógeno o endógeno. La CRH estimula el desdoblamiento de la proopiomelanocortina (POMC), un polipéptido de 241 aminoácidos, por acción de la convertasa de prohormona 1 (PC1, prohormone convertase 1) específica de la hipófisis, lo cual produce ACTH, un péptido de 39 aminoácidos. La ACTH es liberada por las células corticotrópicas de la hipófisis anterior y actúa como el regulador clave de la biosíntesis de cortisol suprarrenal, con otros efectos de corto plazo sobre la biosíntesis de andrógenos suprarrenales y mineralocorticoides. La liberación de CRH, y luego de ACTH, ocurren de manera pulsátil siguiendo un ritmo circadiano bajo el control del hipotálamo, en particular del núcleo supraquiasmático (SCN, suprachiasmatic nucleus), con regulación adicional de una red compleja de genes reloj específicos de cada célula. La liberación circadiana de ACTH la regula en primera instancia la CRH, sin embargo el complejo arginina-vasopresina (AVP) también ejerce una función secretora. Reflejando el patrón de secreción de ACTH, la secreción de cortisol suprarrenal exhibe un ritmo circadiano distinto; comienza a elevarse al amanecer antes de despertar y alcanza niveles máximos en la mañana y concentraciones bajas en la tarde (fig. 3).
En la valoración del eje HPA con pruebas diagnósticas se debe considerar que este es regulado por retroalimentación negativa. Para diagnosticar el exceso de glucocorticoides, se aplica una prueba de supresión con dexametasona. Este fármaco es un potente glucocorticoide sintético que suprime CRH/ACTH uniéndose a receptores de glucocorticoides (GRs, glucocorticoid receptors) hipotalámicos e hipofisarios y, por tanto, inhibe la biosíntesis del cortisol endógeno. Si la producción de cortisol es autónoma (p. ej., un nódulo suprarrenal), la ACTH ya se encuentra suprimida y la dexametasona tiene poco efecto adicional. Si la producción de cortisol la rige un adenoma hipofisario productor de ACTH, la supresión con dexametasona no es eficaz en dosis bajas, pero suele inducir supresión con dosis altas. Si la producción de cortisol es controlada por una fuente ectópica de ACTH, los tumores casi siempre son resistentes a la supresión con dexametasona. Por tanto, esta prueba ayuda a establecer el diagnóstico de síndrome de Cushing y apoyar con el diagnóstico diferencial de exceso de cortisol.
En cambio, para valorar la deficiencia de glucocorticoides, se utiliza la ACTH para estimular la producción de cortisol. La ACTH es un péptido de 39 aminoácidos, pero los primeros 24 son suficientes para producir una respuesta fisiológica. La prueba estándar de estimulación con ACTH consiste en la administración de 0.25 mg de cosintropina (ACTH 1–24) intramuscular (IM) o intravenosa (IV) y la obtención de muestras sanguíneas a los 0, 30 y (de manera opcional) 60 min para cuantificar cortisol. Una respuesta normal se define como una concentración de cortisol > 15 a 20 mcg/100 ml (> 400 a 550 nmol/L), 30 a 60 min después de la estimulación con cosintropina, pero el límite preciso depende del estudio utilizado. Se ha recomendado una versión de esta prueba, pero con una dosis baja (1 mcg de cosintropina IV); sin embargo, no tiene un valor diagnóstico superior y es más complicado llevarla a cabo por la falta de preparaciones de 1 mcg de cosintropina disponibles en el mercado. También puede utilizarse una prueba de tolerancia a la insulina (ITT, insulin tolerance test) para valorar la función suprarrenal. Esta consiste en la inyección de insulina para inducir hipoglucemia, que representa una fuerte señal de tensión que desencadena la liberación de CRH hipotalámica y la activación de todo el eje HPA. La ITT incluye la administración de 0.1 U/kg de insulina regular IV (la dosis debe ser menor si hay probabilidad de hipopituitarismo) y la obtención de muestras sanguíneas a los 0, 30, 60 y 120 minutos para cuantificar glucosa, cortisol y hormona de crecimiento (GH, growth hormone), si también se valora el eje de GH. La glucosa oral o IV se administra después de que el paciente alcanza un estado de hipoglucemia sintomática (por lo general glucosa plasmática < 40 mg/100 ml). Una respuesta normal se define como concentraciones de cortisol > 20 mcg/100 ml y de GH > 5.1 mcg/L; de nuevo, el límite preciso depende del estudio utilizado. La prueba de tolerancia a la insulina exige una cuidadosa vigilancia médica y cuantificaciones secuenciales de glucosa. Está contraindicada en pacientes con coronariopatías, enfermedad cerebrovascular o crisis convulsivas; por lo tanto, la prueba con cosintropina es el estudio de primera línea que se acepta de forma común. La prueba nocturna de metirapona se utiliza de manera alternativa en algunos centros: la metirapona (un fármaco que bloquea la conversión de 11-desoxicortisol en cortisol a través de la 11β-hidroxilasa [véase el tratamiento del síndrome de Cushing]) se administra por vía oral a la medianoche (2500 mg o 30 mg/kg). En individuos sanos, la disminución de cortisol estimula la producción de CRH y ACTH, lo cual incrementa el 11-desoxicortisol. Una respuesta normal consiste en una concentración sérica de 11-desoxicortisol > 287 nmol/L (10 mcg/dl) al iniciar la mañana. La prueba de cortisona salival ambulatoria al despertar como medida de reserva de cortisol suprarrenal es muy prometedora y representa una alternativa potencial no invasiva que puede realizarse en casa.
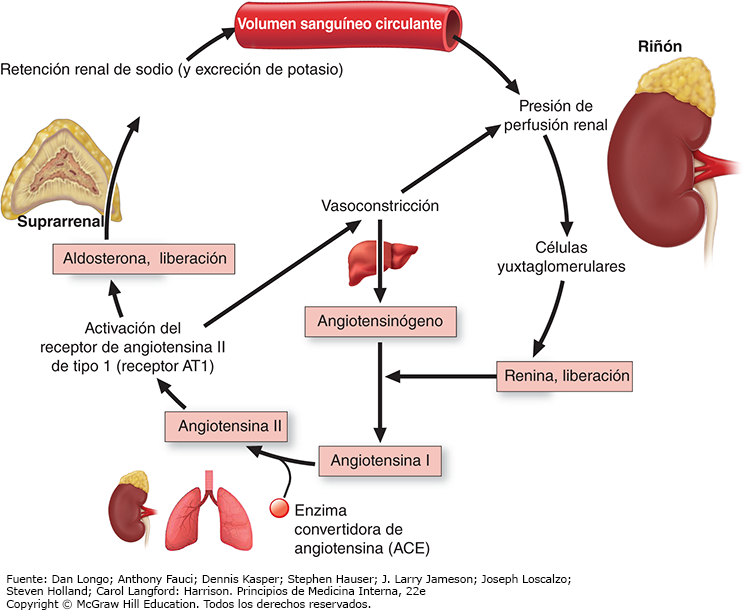
La producción de mineralocorticoides la controla el ciclo regulador de RAA, que inicia con la liberación de la enzima renina de las células yuxtaglomerulares en el riñón, lo que transforma el angiotensinógeno en angiotensina I en el hígado (fig. 4). La enzima convertidora de angiotensina (ACE, angiotensin converting enzyme) desdobla la angiotensina I en angiotensina II, que se une al receptor tipo 1 de angiotensina II y lo activa (receptor AT1 [AT1R, AT1 receptor]), lo cual aumenta la producción de aldosterona suprarrenal y genera vasoconstricción. La aldosterona aumenta la retención de sodio y la excreción de potasio en los riñones, lo cual expande el volumen e incrementa la perfusión renal, que a su vez regula la liberación de renina. Debido a que la síntesis de mineralocorticoides se encuentra en primera instancia bajo control del sistema RAA, el daño hipotalámico-hipofisario no influye de manera significativa en la capacidad de las glándulas suprarrenales para sintetizar aldosterona.
De manera similar al eje HPA, la valoración del sistema RAA puede utilizarse para fines diagnósticos. Si hay exceso de mineralocorticoides, hay una regulación descendente de renina plasmática como mecanismo contrarregulador (véanse más adelante las pruebas diagnósticas). En cambio, en la deficiencia de mineralocorticoides, la renina plasmática está muy aumentada. De manera fisiológica, una carga oral o IV de sodio suprime la aldosterona, una respuesta que se encuentra atenuada o ausente en personas con exceso autónomo de mineralocorticoides.
Síntesis, metabolismo y acción de las hormonas esteroideas
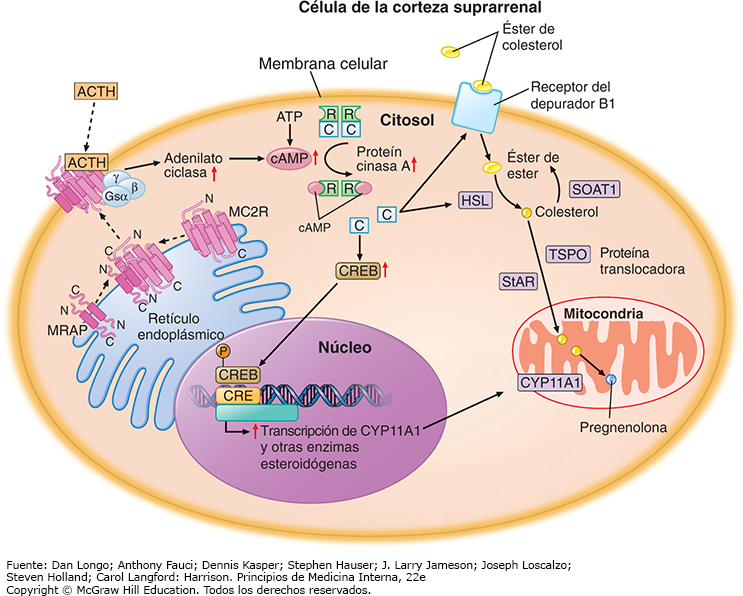
La estimulación de ACTH es necesaria para el inicio de la esteroidogénesis. El receptor de melanocortina 2 (MC2R, melanocortin 2 receptor) que responde a la ACTH, interactúa con la proteína accesoria de MCR2 MRAP, y el complejo se transporta a la membrana de las células corticosuprarrenales, donde se une a la ACTH (fig. 5). La estimulación por ACTH genera AMP cíclico (cAMP), que produce regulación ascendente de la ruta de señalización de la proteincinasa A (PKA, protein kinase A). La PKA inactiva es un tetrámero de dos subunidades reguladoras y dos catalíticas, que se disocia por el cAMP en un dímero de dos subunidades reguladoras unidas al cAMP y dos subunidades catalíticas activas y libres. La activación de la PKA repercute en la esteroidogénesis de tres maneras distintas: 1) aumenta la importación de los esteres de colesterol; 2) incrementa la actividad de la lipasa sensible a hormonas, que desdobla los esteres de colesterol en colesterol para así importarlos al interior de las mitocondrias; y 3) extiende la disponibilidad y la fosforilación de la proteína de unión al elemento de respuesta al cAMP (CREB, cAMP response element binding), un factor transcripcional que aumenta la transcripción de CYP11A1 y de otras enzimas necesarias para la biosíntesis de glucocorticoides.
La esteroidogénesis suprarrenal ocurre en sitios específicos; la síntesis de mineralocorticoides sucede en la zona glomerulosa externa, la producción de glucocorticoides en la zona fasciculada y los andrógenos suprarrenales se generan en la zona reticular interna, sirviendo como precursores para los andrógenos 11-oxigenados y los clásicos (fig. 1). Todas las rutas de la esteroidogénesis necesitan la entrada de colesterol a las mitocondrias, un proceso que inicia por la acción de la proteína reguladora de la esteroidogénesis aguda (StAR, steroidogenic acute regulatory protein), que transporta el colesterol de la membrana mitocondrial externa a la membrana mitocondrial interna. La mayoría de las enzimas de la esteroidogénesis pertenece a la familia del citocromo P450 (CYP), que se localizan en la mitocondria (enzima proteolítica de la cadena lateral, CYP11A1; 11β-hidroxilasa, CYP11B1; sintasa de aldosterona y CYP11B2) o en la membrana del retículo endoplásmico (17α-hidroxilasa, CYP17A1; 21-hidroxilasa, CYP21A2; aromatasa y CYP19A1). Estas enzimas necesitan donación de electrones por medio de cofactores enzimáticos redox específicos, P450 oxidorreductasa (POR, P450 oxidoreductase) y adrenodoxina/reductasa adrenodoxina (ADX/ADR) para las enzimas CYP microsómicas y mitocondriales, en dicho orden. Además, la deshidrogenasa de 3β-hidroxiesteroide tipo 2 (3β-HSD2, 3β-hydroxysteroid dehydrogenase type 2) (una deshidrogenasa de cadena corta), también llamada Δ4,Δ5 isomerasa, desempeña una función clave en la esteroidogénesis suprarrenal
La enzima que desdobla la cadena lateral de colesterol, CYP11A1, produce pregnenolona. La síntesis de glucocorticoides requiere la biotransformación de pregnenolona en progesterona por medio de la 3β-HSD2, seguida de biotransformación en 17-hidroxiprogesterona (17OHP) por CYP17A1, hidroxilación en el carbono 21 a través de CYP21A2 y, al final, 11β-hidroxilación por medio de CYP11B1 para producir cortisol activo (fig. 1). La síntesis de mineralocorticoides también necesita progesterona, que al inicio se transforma en desoxicorticosterona (DOC) por la CYP21A2, y luego se transforma por medio de corticosterona y 18-hidroxicorticosterona en aldosterona en tres pasos catalizados por CYP11B2. Para la síntesis de andrógenos suprarrenales, la pregnenolona experimenta biotransformación por CYP17A1, que de manera excepcional cataliza dos reacciones enzimáticas. Por medio de su actividad 17α-hidroxilasa, CYP17A1 transforma la pregnenolona en 17-hidroxipregnenolona, seguida de la generación del precursor universal de esteroides sexuales, la DHEA, mediante la actividad 17,20 liasa de CYP17A1. La mayor parte de la DHEA es secretada por las glándulas suprarrenales en forma de su sulfato ester, DHEAS, producido por la sulfotransferasa de DHEA (SULT2A1). La DHEA se convierte en androstenediona, que se activa para formar testosterona o se canaliza en la ruta de los andrógenos 11-oxigenados mediante 11β-hidroxilación (CYP11B1).
Después de su liberación de las glándulas suprarrenales, el cortisol circula en el torrente sanguíneo unido en primera instancia a la globulina transportadora de cortisol (CBG, cortisol-binding globulin) y, en menor grado, a la albúmina (solo una pequeña fracción circula como hormona libre no unida). Se considera que el cortisol libre entra de manera directa a las células, sin necesitar de transporte activo. Además, en muchos tejidos periféricos efectores que responden a los glucocorticoides, como el tejido adiposo, el hígado, los músculos y el cerebro, el cortisol se genera a partir de cortisona inactiva dentro de la célula por medio de la enzima 11β-hidroxiesteroide deshidrogenasa de tipo 1 (11β-HSD1, 11β-hydroxysteroid dehydrogenase type 1) (fig. 6). En consecuencia, la 11β-HSD1 funciona como un pre-receptor regulador de la acción de los glucocorticoides, que es específico de cada tejido. Para la transformación de cortisona inactiva en cortisol activo, la 11β-HSD1 necesita fosfato de dinucleótido de nicotinamida y adenina (NADPH, nicotinamide adenine dinucleotide phosphate [en su forma reducida]), el cual lo proporciona la enzima hexosa-6-fosfato deshidrogenasa (H6PDH, hexose-6-phosphate dehydrogenase). Al igual que el dominio catalítico de 11β-HSD1, la H6PDH se localiza en la luz del retículo endoplásmico y biotransforma glucosa-6-fosfato (G6P, glucose-6-phosphate) en 6-fosfogluconato (6PGL, 6-phosphogluconate), y de ese modo regenera el NADP+ en NADPH, que impulsa la activación del cortisol a partir de cortisona por medio de la 11β-HSD1.
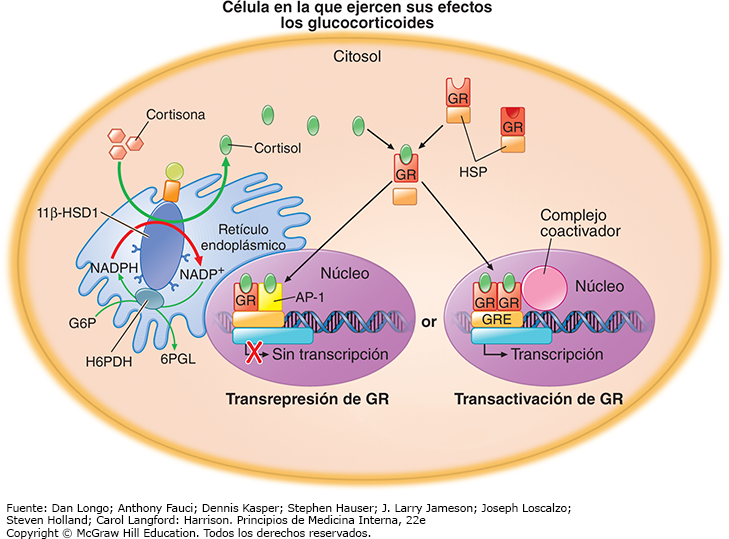
En el citosol de células efectoras, el cortisol se une al GR y lo activa, lo cual disocia las proteínas de choque térmico (HSP, heat shock proteins) del receptor y ocurre dimerización subsecuente (fig. 6). Los dímeros del GR unidos al cortisol se traslocan al núcleo y activan los elementos de respuesta a glucocorticoides (GRE, glucocorticoid response element) en la secuencia de ácido desoxirribonucleico (DNA, deoxyribonucleic acid) y de esta manera aumentan la transcripción de los genes regulados por glucocorticoides (transactivación de GR). Sin embargo, el GR unido al cortisol también puede formar heterodímeros con factores de transcripción como AP-1 o el factor nuclear-κB (NF-κB, nuclear factor-κB), lo que da como resultado la trans-represión de genes proinflamatorios, un mecanismo de gran importancia para la acción antiinflamatoria de los glucocorticoides. Hay que destacar que la corticosterona también tiene actividad glucocorticoide, aunque mucho más leve que el cortisol mismo. Sin embargo, en los roedores, la corticosterona es el principal glucocorticoide, y en pacientes con deficiencia de 17-hidroxilasa, la falta de cortisol puede ser compensada por las concentraciones altas de corticosterona que se acumulan como consecuencia del bloqueo enzimático.
La enzima microsómica 11β-hidroxiesteroide deshidrogenasa tipo 2 (11β-HSD2) inactiva el cortisol y lo transforma en corticosterona (fig. 7), de manera predominante en los riñones, pero también en el colon, las glándulas salivales y otros tejidos focalizados por los mineralocorticoides. El cortisol y la aldosterona se unen al receptor de mineralocorticoides (MR, mineralocorticoid receptor) con igual afinidad; sin embargo, el cortisol circula por el torrente sanguíneo en una concentración casi 1 000 veces superior. Por tanto, solo la inactivación rápida de cortisol a cortisona por la 11β-HSD2 previene la activación de MR por el exceso de cortisol, y de esta manera actúa como un modulador de la ruta del MR en tejidos específicos. Además del cortisol y la aldosterona, la desoxicorticosterona (DOC) (fig. 1) también ejerce actividad mineralocorticoide. La acumulación de DOC por deficiencia de 11β-hidroxilasa o por la producción excesiva relacionada con un tumor puede resultar en el exceso de mineralocorticoides.
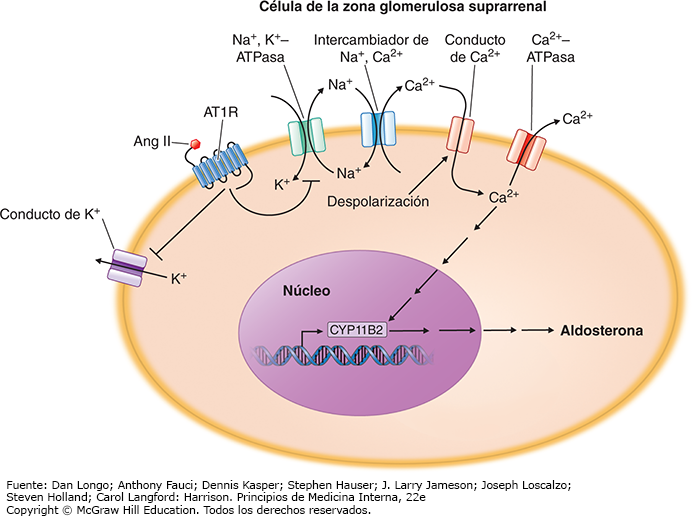
La biosíntesis de aldosterona en las células de la zona glomerulosa suprarrenal es dirigida por la enzima sintasa de aldosterona (CYP11B2). La unión de angiotensina II al receptor AT1 ocasiona despolarización de la membrana de las células glomerulosas al incrementar el sodio intracelular por la inhibición de las enzimas sodio-potasio (Na+/K+) ATPasas, así como de los conductos del potasio. Esto impulsa un aumento del calcio intracelular porque se abren los conductos de calcio dependientes de voltaje o se inhiben las enzimas calcio (Ca2+) ATPasas. En consecuencia, se activa la ruta de señalización de calcio, lo que resulta en una regulación ascendente de la transcripción de CYP11B2 (fig. 8).
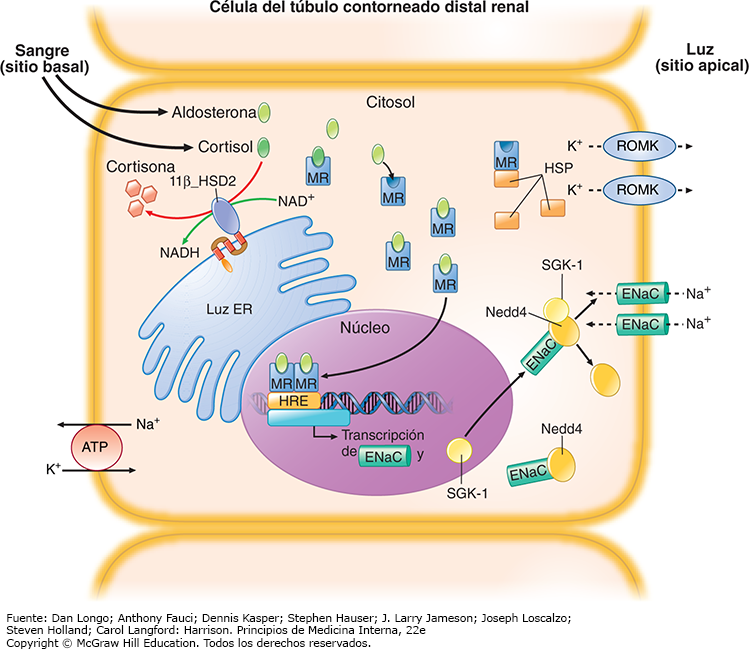
De manera análoga a la acción del cortisol por la vía del GR, la unión de aldosterona (o cortisol) a MR en las células de los túbulos renales disocia los complejos HSP-receptor, lo que permite la homodimerización de los MR y la traslocación de las hormonas unidas a los dímeros MR hacia el núcleo (fig. 7). El receptor de mineralocorticoides activado aumenta la transcripción del conducto epitelial de sodio (ENaC, epithelial sodium channel) y de la quinasa 1 sérica inducible por glucocorticoides (SGK-1, serum glucocorticoid inducible kinase 1). En el citosol, la interacción entre ENaC y Nedd4 previene la expresión del ENaC en la superficie celular. Sin embargo, la SGK-1 fosforila los residuos de serina en la proteína Nedd4, reduce la interacción entre Nedd4 y ENaC, y, en consecuencia, aumenta el paso de ENaC a la superficie celular, donde media la retención de sodio dependiente de la extrusión de potasio a través del conducto de potasio de la corteza medular externa (ROMK, renal outer medullary potassium).
Síndrome de Cushing
El síndrome de Cushing refleja una constelación de manifestaciones clínicas que son resultado de la exposición crónica al exceso de glucocorticoides por cualquier etiología. La enfermedad puede ser dependiente de ACTH (p. ej., adenoma hipofisario corticotropo o secreción ectópica de ACTH por tumores no hipofisarios), independiente de ACTH (p. ej., adenoma corticosuprarrenal, carcinoma corticosuprarrenal [ACC, adrenocortical carcinoma] o hiperplasia suprarrenal nodular) o yatrogénica (p. ej., administración de glucocorticoides exógenos para tratar diversas enfermedades inflamatorias). El término enfermedad de Cushing se refiere de manera específica al síndrome de Cushing ocasionado por un adenoma hipofisario corticotropo.
Epidemiología
El síndrome de Cushing yatrógeno (o exógeno) es por mucho la causa más común, considerando que casi 1% de la población utiliza de forma crónica tratamientos de glucocorticoides por sus propiedades antiinflamatorias e inmunosupresoras. Por el contrario, el síndrome de Cushing endógeno se considera, en general, una enfermedad inusual; su incidencia es 1.8 a 3.2 casos por millón de personas al año. Sin embargo, es motivo de debate si el exceso leve de cortisol es más prevalente en pacientes con características de Cushing, como obesidad centrípeta, diabetes tipo 2 y fracturas vertebrales osteoporóticas, reconociendo que estas son relativamente inespecíficas y comunes en la población.
Etiología
En la inmensa mayoría de pacientes con síndrome de Cushing endógeno, la causa subyacente es un tumor neuroendocrino hipofisario productor de ACTH, es decir, un adenoma corticotropo (Cuadro 1), como lo describió por primera vez en 1912 Harvey Cushing. La enfermedad de Cushing afecta con más frecuencia a las mujeres, excepto en los casos prepuberales, donde es más común en niños de sexo masculino. En cambio, el síndrome de secreción ectópica de ACTH de se identifica más a menudo en varones. Solo 10% de los pacientes con síndrome de Cushing tiene una causa suprarrenal primaria (es decir, exceso de cortisol autónomo, independiente de ACTH) y la mayoría de estos pacientes son mujeres.
|
|
MUJER:VARÓN |
% |
|
Síndrome de Cushing dependiente de ACTH |
|
90 |
|
Enfermedad de Cushing (= adenoma hipofisario productor de ACTH) |
4:1 |
75 |
|
Síndrome de secreción ectópica de ACTH (por secreción de ACTH por tumores carcinoides de páncreas o bronquios, cáncer pulmonar microcítico, carcinoma medular de tiroides, feocromocitoma y otros) |
1:1 |
15 |
|
Síndrome de Cushing no dependiente de ACTH |
4:1 |
10 |
|
Adenoma corticosuprarrenal |
|
5-10 |
|
Carcinoma corticosuprarrenal |
|
1 |
|
Causas poco frecuentes: hiperplasia suprarrenal macronodular, enfermedad suprarrenal nodular pigmentada primaria (micronodular, macronodular, o ambos); síndrome de McCune-Albright |
|
<1 |
Cuadro 1. Causas del síndrome de Cushing endógeno.
El exceso de ACTH, en al menos el 90% de los pacientes con enfermedad de Cushing, es ocasionado por un microadenoma hipofisario corticotropo, con frecuencia de solo pocos milímetros de diámetro. Los macroadenomas hipofisarios (tumores > 1 cm de tamaño) se identifican solo en 5% a 10% de los pacientes, pueden tener características invasivas y afectar el quiasma óptico y los senos cavernosos. Los adenomas hipofisarios corticotropos se observan en forma esporádica; sin embargo, en ocasiones excepcionales pueden identificarse en casos de neoplasia endocrina múltiple tipo 1 (MEN1, multiple endocrine neoplasia type 1), MEN 4, adenomas hipofisarios aislados familiares, complejo de Carney, síndrome DICER1, y síndrome de Lynch. Los adenomas hipofisarios que causan enfermedad de Cushing con frecuencia portan variantes somáticas activadoras en el gen USP8, que codifica la enzima desubicuitinadora denominada proteasa 8 específica de ubicuitina, lo que conduce a la activación constitutiva de la señalización del factor de crecimiento epidérmico (EGF, epidermal growth factor) y, en consecuencia, a la expresión ascendente de POMC, precursora de ACTH. Las mutaciones en USP8 se encuentran en 11% a 62% de los adenomas corticotropos y con mayor frecuencia en adultos y mujeres con enfermedad de Cushing. De manera menos frecuente, variantes somáticas del gen receptor de glucocorticoides (NR3C1), BRAF, USP48 y TP53 se observan en el tejido tumoral de casos de enfermedad de Cushing con USP8 de tipo nativo.
La producción ectópica de ACTH es ocasionada de manera predominante por tumores carcinoides ocultos, con mayor frecuencia en los pulmones, pero también se pueden localizar en el timo o en el páncreas. A causa de su tamaño pequeño, estos tumores suelen ser difíciles de localizar. El cáncer pulmonar microcítico avanzado puede ocasionar producción ectópica de ACTH. En casos excepcionales se ha descubierto que la producción ectópica de CRH y/o ACTH proviene de un carcinoma medular de la tiroides o de un feocromocitoma; este último secreta catecolaminas y ACTH de forma simultánea.
La mayoría de los pacientes con exceso de cortisol endógeno independiente de ACTH alberga un adenoma suprarrenal productor de cortisol y se han identificado mutaciones somáticas activadoras en el gen PRKACA (subunidad catalítica de la PKA) como causa de la enfermedad en 40% de estos tumores. Se han observado con menor frecuencia defectos somáticos inactivadores en PRKAR1A (que codifica una de las unidades reguladoras de la PKA) y en GNAS (que codifica la subunidad estimuladora α 1 de la proteína G, GNAS-1 [polipéptido 1 estimulador de la actividad de α de la proteína de unión al nucleótido de guanina]) en adenomas un unilaterales productores de cortisol. Los ACC también pueden generar exceso de cortisol independiente de ACTH y a menudo son grandes, con una producción excesiva de varias clases de corticoesteroides.
Una causa inusual pero notable de exceso de cortisol suprarrenal es la hiperplasia suprarrenal macronodular bilateral primaria (PBMAH, primary bilateral macronodular adrenal hyperplasia) con ACTH circulante baja, pero con evidencia de estimulación autocrina para la producción de cortisol mediante la generación de ACTH intra-suprarrenal. Estos nódulos hiperplásicos también se caracterizan por la expresión ectópica de receptores acoplados a proteína G que no se encuentran de manera normal en la glándula suprarrenal, incluyendo receptores para hormona luteinizante, vasopresina, serotonina, interleucina-1, catecolaminas o péptido inhibidor gástrico (GIP, gastric inhibitory peptide), la causa de síndrome de Cushing dependiente de alimentos. La activación de estos receptores incrementa la señalización de PKA, como ocurre de manera fisiológica con la ACTH, con un aumento subsecuente de la producción de cortisol. Se ha identificado una combinación de mutaciones somáticas y de la línea germinal en el gen supresor de tumores ARMC5 como causa prevalente de enfermedad de Cushing por hiperplasia suprarrenal macronodular bilateral; estos pacientes a menudo manifiestan evidencias bioquímicas de síndrome de Cushing, pero sin signos clínicos específicos, los cuales aparecen de manera lenta en el transcurso de décadas y aceleran el riesgo cardiovascular. Mutaciones activadoras constitutivas en el gen PRKACA y variantes inactivadoras en el gen KDM1A (desmetilasa 1A de histonas específica de lisinas) también pueden ser causas inusuales de hiperplasia suprarrenal macronodular bilateral; las últimas se vinculan con el síndrome de Cushing dependiente GIP. La hiperplasia suprarrenal macronodular bilateral con exceso de cortisol también se ha descrito en otros síndromes familiares: MEN1, leiomiomatosis hereditaria y cáncer de células renales (gen FH) y poliposis adenomatosa familiar (gen APC).
Las mutaciones de línea germinal en el gen PRKAR1A se identifican en pacientes con enfermedad suprarrenal nodular pigmentada primaria (PPNAD, primary pigmented nodular adrenal disease) como parte del complejo de Carney, una condición caracterizada por neoplasia múltiple autosómica dominante asociada a mixomas cardiacos y cutáneos, hiperlentiginosis, tumores de células de Sertoli, tumores ováricos, tumores mamarios, nódulos tiroideos, Schwannomas y PPNAD. La PPNAD puede presentarse como hiperplasia micronodular o macronodular, o ambas. Las fosfodiesterasas influyen en el cAMP intracelular y, por lo tanto, pueden determinar la activación de la PKA. Se han identificado mutaciones en PDE11A y PDE8B en pacientes con hiperplasia suprarrenal bilateral y Cushing con o sin evidencias de PPNAD.
Otra causa poco frecuente de síndrome de Cushing independiente de ACTH es el síndrome de McCune-Albright, que también se vincula con displasia fibrosa poliostótica, manchas de café con leche unilaterales y pubertad precoz. El síndrome de McCune-Albright es ocasionado por mutaciones activadoras en el gen GNAS (cuadro 1).
Manifestaciones clínicas
Los glucocorticoides afectan casi a todas las células del cuerpo y, por lo tanto, los signos de exceso de cortisol afectan a múltiples sistemas fisiológicos (cuadro 2), con incremento regulatorio de la gluconeogénesis, la lipólisis y el catabolismo de las proteínas, lo que determina las características más prominentes. Además, el exceso de secreción de glucocorticoides supera la capacidad de la 11β-HSD2 para inactivar con rapidez el cortisol y transformarlo en cortisona en el riñón, y de este modo realiza acciones mineralocorticoides, manifestadas como hipertensión, hipopotasemia y edema. El exceso de glucocorticoides también interfiere en los sistemas regulatorios centrales, lo que ocasiona la supresión de gonadotropinas con hipogonadismo y amenorrea subsiguientes e inhibición del eje hipotálamo-hipófisis-tiroides, lo cual disminuye la secreción de hormona estimulante de la tiroides (TSH, thyroid-stimulating hormone).
|
SISTEMA O COMPARTIMIENTO CORPORAL |
SIGNOS Y SÍNTOMAS |
|
Grasa corporal |
Aumento de peso, obesidad central, cara redonda y tejido adiposo en la parte posterior del cuello (“giba de búfalo”) |
|
Piel |
Rubicundez facial, piel delgada y brillante, facilidad para desarrollar hematomas, estrías amplias y de color púrpura, acné e hirsutismo |
|
Huesos |
Osteopenia, osteoporosis (fracturas vertebrales) y disminución del crecimiento lineal en niños |
|
Músculos |
Debilidad, miopatía proximal (atrofia prominente de los músculos superiores de la pierna y de los glúteos, con dificultad para subir escaleras o levantarse de una silla) |
|
Aparato cardiovascular |
Hipertensión, hipopotasemia, edema y aterosclerosis |
|
Metabolismo |
Intolerancia a la glucosa/diabetes y dislipidemia |
|
Aparato reproductor |
Disminución de la libido; en mujeres, amenorrea (por inhibición de la liberación de gonadotropinas mediada por cortisol) |
|
Sistema nervioso central |
Irritabilidad, labilidad emocional, depresión, insomnio y perturbaciones del sueño, en ocasiones defectos cognitivos; en casos graves, sicosis paranoide |
|
Sangre y sistema inmunitario |
Mayor susceptibilidad a infecciones, aumento del recuento de leucocitos, eosinopenia, hipercoagulabilidad con mayor riesgo de trombosis venosa profunda y embolia pulmonar |
Cuadro 2. Signos y síntomas del síndrome de Cushing.
Muchos de los signos y síntomas clínicos observados en el síndrome de Cushing son inespecíficos en términos relativos e incluyen manifestaciones como obesidad, diabetes, hipertensión diastólica, hirsutismo y depresión, que se observan con frecuencia en personas que no tienen síndrome de Cushing. En consecuencia, la valoración clínica minuciosa es fundamental cuando se evalúan casos sospechosos. Se debe sospechar síndrome de Cushing cuando en un mismo paciente se identifican varias características clínicas, en particular cuando se encuentran hallazgos más específicos o si se manifiestan a una edad inusual, por ejemplo, osteoporosis en un paciente joven. Estos incluyen fragilidad de la piel, facilidad para desarrollar hematomas amplios (> 1 cm), estrías violáceas (fig. 9) y signos de miopatía proximal, que son más evidentes cuando la persona intenta levantarse de una silla sin el apoyo de las manos o cuando sube escaleras. Las manifestaciones clínicas del síndrome de Cushing no difieren mucho entre las diferentes etiologías. En el síndrome de secreción ectópica de ACTH puede observarse hiperpigmentación en los nudillos, cicatrices o áreas de la piel expuestas a mucha fricción (fig. 9), por los efectos estimuladores del exceso de ACTH y otros productos del desdoblamiento de la POMC sobre la producción de pigmentos de melanocitos. Además, individuos con síndrome de secreción ectópica de ACTH, y algunos sujetos con carcinoma corticosuprarrenal como causa de Cushing, tienen un inicio más enérgico y una progresión rápida de los signos y síntomas clínicos, en particular del edema, la hipopotasemia y la hipertensión.
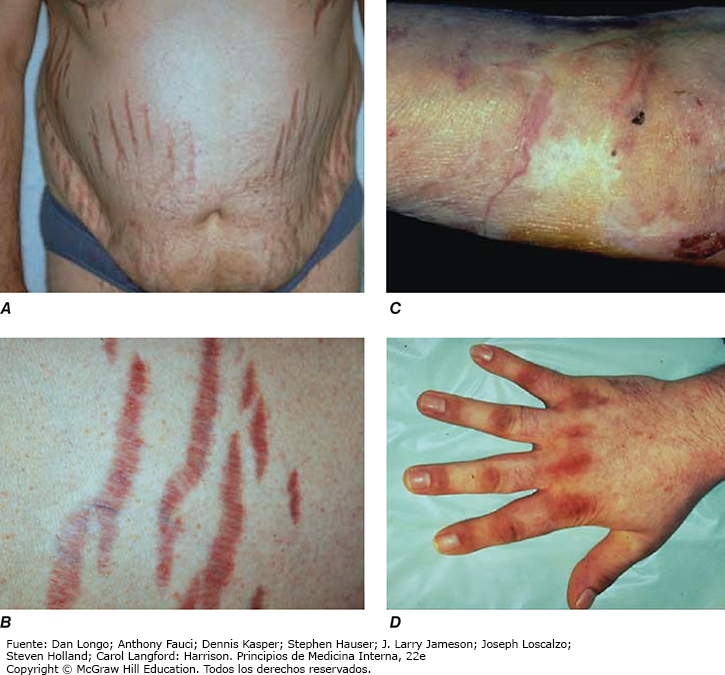
Los sujetos con síndrome de Cushing pueden estar en gran peligro de desarrollar trombosis venosas profundas, con aumento del riesgo de embolia pulmonar por el estado de hipercoagulabilidad vinculado con el exceso de cortisol. La mayoría de los individuos también experimenta síntomas siquiátricos, en general en forma de ansiedad o depresión; sin embargo, también pueden presentarse sicosis depresiva o paranoide aguda. Aun después de la curación, la salud de largo plazo puede verse afectada por deterioro persistente de la calidad de vida vinculado a la salud e incremento del riesgo de padecer enfermedades cardiovasculares y osteoporosis con fracturas vertebrales, lo cual depende de la duración y el grado de exposición al exceso significativo de cortisol.
Diagnóstico
El primer paso más importante del tratamiento de los pacientes con sospecha de síndrome de Cushing es establecer el diagnóstico correcto. La mayor parte de los errores en la valoración clínica, que conducen a estudios de imagen o a cirugías innecesarios, se debe a que no se sigue el protocolo diagnóstico (fig. 10). Este protocolo requiere establecer el diagnóstico de síndrome de Cushing con toda certeza antes de aplicar cualquier prueba para el diagnóstico diferencial de la enfermedad. En un principio, después de excluir el uso de glucocorticoides exógenos como la causa de los signos y síntomas clínicos, los casos sospechosos deben estudiarse si hay múltiples manifestaciones progresivas de síndrome de Cushing, en particular hallazgos con un valor de discriminación potencialmente alto. También se indica excluir el exceso de cortisol en pacientes con masas suprarrenales descubiertas de manera incidental.
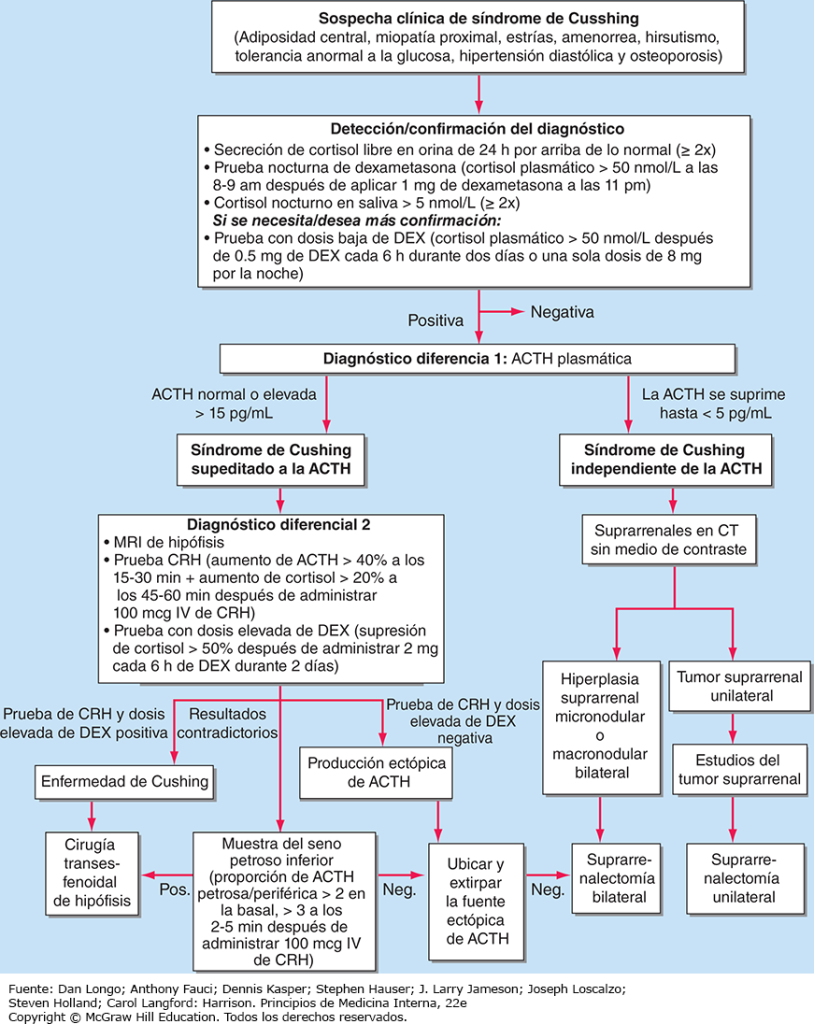
El diagnóstico de Cushing puede considerarse confirmado cuando los resultados de varias pruebas son consistentes con dicho trastorno. Estos estudios pueden incluir aumento de la excreción de cortisol libre en orina de 24 h en tres recolecciones separadas, incapacidad para suprimir de manera apropiada el cortisol sérico matutino después de la exposición nocturna a dexametasona y evidencias de pérdida de secreción de cortisol diurno con concentraciones altas de cortisol sérico o salival a la medianoche, el momento de menor secreción fisiológica (fig. 10). Deben excluirse los factores que pueden afectar el resultado de estas pruebas diagnósticas, como la obtención incompleta de orina en 24 horas o la inactivación rápida de la dexametasona por el consumo simultáneo de fármacos que inducen al CYP3A4 (p. ej., antiepilépticos y rifampicina). El consumo simultáneo de anticonceptivos orales que aumentan la CBG y, por tanto, el cortisol total, puede ocasionar falta de supresión después de la administración de dexametasona. Si se tienen dudas, las pruebas deben repetirse después de cuatro a seis semanas de haber interrumpido los estrógenos orales. Los pacientes con estados de seudo-Cushing (p. ej., hipercortisolismo no neoplásico vinculado con el consumo de alcohol o relacionado con depresión mayor u obesidad mórbida) o con síndrome de Cushing cíclico pueden necesitar más pruebas para confirmar o excluir de manera segura el diagnóstico de síndrome de Cushing. Además, los análisis bioquímicos utilizados pueden afectar los resultados de los estudios, puesto que la especificidad constituye un problema frecuente en los análisis basados en anticuerpos para la cuantificación de cortisol libre urinario. Estos análisis han mejorado en gran medida con la introducción de la espectrometría de masas en tándem de alta especificidad.
Diagnóstico diferencial
La valoración de pacientes con síndrome de Cushing confirmado la debe ejecutar un endocrinólogo e inicia con el diagnóstico diferencial de exceso de cortisol dependiente de ACTH e independiente de ACTH (fig. 10). En general, las concentraciones plasmáticas de ACTH están suprimidas en casos de exceso de cortisol suprarrenal autónomo, como consecuencia del aumento de la retroalimentación negativa al hipotálamo y la hipófisis. En cambio, los pacientes con síndrome de Cushing dependiente de ACTH tienen ACTH normal o alta en el plasma; se observan concentraciones muy altas en algunos pacientes con síndrome de secreción ectópica de ACTH. Es importante mencionar que los estudios de imagen solo deben solicitarse después de establecer si el exceso de cortisol depende o no de la ACTH, debido a que la presencia de nódulos en la hipófisis o las glándulas suprarrenales es un hallazgo común en la población general. En pacientes con exceso de cortisol independiente de ACTH confirmado, se indican estudios de imagen de las glándulas suprarrenales (fig. 11), de preferencia una tomografía computarizada (CT, computed tomography) sin contraste. Esto permite valorar la morfología de las glándulas suprarrenales y determinar la densidad de los tumores, es decir, el valor de atenuación medido en unidades Hounsfield (HU, Hounsfield Units), lo cual ayuda a distinguir entre lesiones suprarrenales malignas y benignas (nótese que esto solo aplica a CT sin contraste; las CT con medio de contraste no generan esta información).
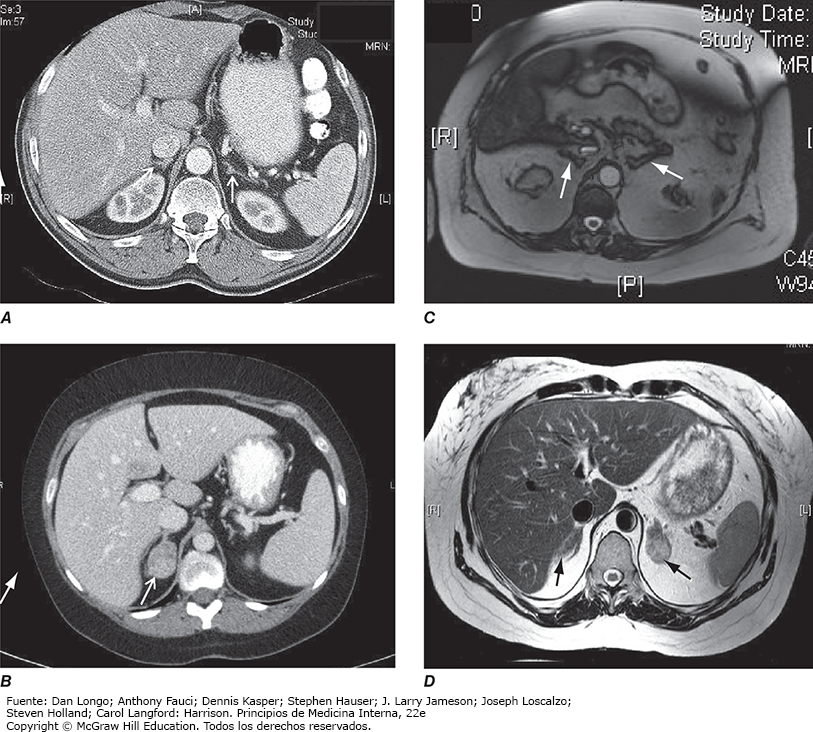
La imagen de resonancia magnética (MRI, magnetic resonance imaging) de la hipófisis es el estudio preferido en casos de hipercortisolismo dependiente de ACTH; sin embargo, esta técnica no permite identificar alteraciones hasta en 40% de los casos porque los tumores pequeños se encuentran por debajo de la sensibilidad de detección. De manera característica, los adenomas hipofisarios corticotropos no exhiben intensificación de contraste después de la administración de gadolinio en las MRI ponderadas en T1. En todos los casos confirmados de síndrome de Cushing dependiente de ACTH se necesitan más pruebas para el diagnóstico diferencial de enfermedad de Cushing hipofisaria y síndrome de secreción ectópica de ACTH. Estas pruebas aprovechan el hecho de que la mayor parte de los adenomas corticotropos hipofisarios aún presentan características reguladoras, que incluyen la supresión residual de ACTH por dosis altas de glucocorticoides y la capacidad de respuesta a CRH o desmopresina, un análogo sintético de la vasopresina. Por el contrario, las fuentes ectópicas de ACTH suelen ser resistentes a la supresión con dexametasona y no responden a CRH o desmopresina (fig. 10). Sin embargo, debe destacarse que una pequeña minoría de tumores ectópicos productores de ACTH presentan respuestas dinámicas similares a las de los tumores corticotropos hipofisarios. Si ambas pruebas muestran resultados discordantes o si hay alguna otra razón para dudar, el diagnóstico diferencial puede aclararse con la obtención de muestras bilaterales del seno petroso inferior (IPSS, inferior petrosal sinus sampling) y muestras concurrentes de sangre para ACTH en el seno petroso inferior derecho e izquierdo y una vena periférica. Una taza basal aumentada de ACTH plasmática central/ACTH periférica > 2 y > 3 2–5 min después de la inyección de CRH indica enfermedad de Cushing (fig. 10) con sensibilidad y especificidad muy altas. Debe destacarse que los resultados de la IPSS no son confiables para lateralización (predicción de la localización del tumor dentro de la hipófisis), porque hay una amplia variabilidad entre individuos en el drenaje venoso de la región hipofisaria. Es importante no administrar fármacos que disminuyan las concentraciones de cortisol antes de las pruebas de IPSS.
Si las pruebas diagnósticas diferenciales indican síndrome de secreción ectópica de ACTH, entonces los estudios de imagen deben incluir CT de cortes finos y de alta resolución del tórax y el abdomen para explorar los pulmones, el timo y el páncreas. Si no se identifican lesiones, puede considerarse una MRI del tórax, porque los tumores carcinoides por lo general muestran alta intensidad de señal en imágenes ponderadas en T2. Además, la gammagrafía con octreótido y la tomografía por emisión de positrones (PET)/CT con 68Ga DOTATATE pueden ser útiles en algunos casos debido a que los tumores productores de ACTH ectópica a menudo expresan receptores de somatostatina. Según la causa sospechada, en los pacientes con síndrome de secreción ectópica de ACTH también deben obtenerse muestras sanguíneas para cuantificar hormonas intestinales en ayuno, cromogranina A y calcitonina y para excluir feocromocitoma con datos bioquímicos.
Tratamiento de Síndrome de Cushing
El síndrome de Cushing manifiesto conlleva un mal pronóstico si no se trata. En la enfermedad independiente de ACTH, el tratamiento consiste en la extirpación quirúrgica del tumor suprarrenal o la hiperplasia nodular. Para tumores benignos puede utilizarse un acceso de mínima invasión, en tanto que para tumores más grandes y en los que se sospeche malignidad, se prefiere un acceso abierto.
En la enfermedad de Cushing, el tratamiento preferido es la extirpación selectiva del tumor hipofisario corticotropo, en general por una vía de acceso transesfenoidal endoscópica. Esto resulta en una tasa inicial de curación de 60 a 80% cuando la realiza un cirujano con mucha experiencia, aunque los índices de remisión son mucho más bajos (12% a 70%) en pacientes que presentan tumores más grandes o invasivos. Sin embargo, aun después de la remisión inicial luego de la cirugía, la vigilancia a largo plazo es importante porque puede haber una recaída tardía en un número significativo de individuos. Si la enfermedad hipofisaria persiste después de la cirugía o recurre después de la remisión inicial, hay varias opciones terapéuticas, como una segunda cirugía, radioterapia fraccionada, radiocirugía estereotáctica, tratamiento farmacológico y suprarrenalectomía bilateral. Estas opciones deben aplicarse de manera individual.
En algunos pacientes con síndrome de Cushing manifiesto y grave (p. ej., dificultad para controlar hipertensión hipopotasémica, sicosis aguda o infecciones que ponen en riesgo la vida), es necesario iniciar tratamiento médico para controlar pronto el exceso de cortisol durante el periodo previo a la cirugía, lo cual también ayuda a mitigar la hipercoagulación y, por lo tanto, el riesgo quirúrgico. De manera similar, las personas con metástasis de carcinomas productores de glucocorticoides pueden necesitar tratamiento con fármacos anti-glucocorticoides a largo plazo. En casos de síndrome de secreción ectópica de ACTH en los que el tumor no puede localizarse, es necesario ponderar con mucha precaución si el tratamiento farmacológico o la suprarrenalectomía bilateral es la opción más adecuada; esta última facilita la cura inmediata, pero es indispensable la restitución de corticoesteroides de por vida. En esta situación es fundamental asegurar la vigilancia regular con estudios de imagen para identificar la fuente de ACTH ectópica.
Los fármacos orales con eficacia establecida para tratar el síndrome de Cushing son el osilodrostat, la metirapona, el ketoconazol, el levoketoconazol y la mifepristona. El osilodrostat y la metirapona inhiben la síntesis de cortisol en el nivel de la 11β-hidroxilasa (fig. 1). El antimicótico ketoconazol y su estereoisómero levoketoconazol inhiben los pasos iniciales de la esteroidogénesis. La mifepristona es un bloqueador de GR, y por lo tanto mitiga los efectos del exceso de cortisol en tejidos focalizados. Las dosis iniciales típicas son 1 a 2 mg cada 12 h de osilodrostat (dosis diaria máxima [DDM], 60 mg), 500 mg cada 8 h de metirapona (DDM, 6 g), 200 mg cada 8 h de ketoconazol (DDM, 1 200 mg), 150 mg cada 12 h de levoketoconazol (DDM, 1200 mg) y 300 mg de mifepristona (DDM, 1200 mg). El mitotano, un derivado del insecticida o,p′DDD, es un agente suprarrenolítico que también es eficaz para reducir el cortisol. Por su perfil de efectos secundarios, se utiliza con más frecuencia en casos de ACC, pero también se ha utilizado el tratamiento con dosis bajas (500–1000 mg por día) en el síndrome de Cushing benigno. En casos graves de exceso de cortisol puede administrarse etomidato, un agente que bloquea de modo potente la 11β-hidroxilasa y la sintasa de aldosterona, para disminuir con rapidez el cortisol. Este fármaco se administra a través de goteo IV continuo en dosis bajas, no anestésicas. Para la enfermedad de Cushing, la administración subcutánea o IM de pasireotida, un agonista del receptor de somatostatina, constituye otra opción terapéutica cuando no es posible lograr la curación quirúrgica. La cabergolina, un agonista de los receptores de dopamina, también se utiliza fuera de sus aplicaciones autorizadas para tratar a ciertos pacientes con enfermedad de Cushing, en especial a aquellos con exceso leve de cortisol, tumor residual, o ambos, por su potencial para reducir los tumores.
Después de la extirpación exitosa de un tumor productor de cortisol o ACTH, el eje HPA permanece suprimido. Por lo tanto, es necesario iniciar la restitución con hidrocortisona al momento de la cirugía y disminuirla con lentitud después de la recuperación, para permitir la adaptación fisiológica a las concentraciones normales de cortisol. Según el grado y la duración del exceso de cortisol, el eje HPA puede necesitar de muchos meses o incluso años para recuperar su función normal, y en ocasiones no se recupera. En general, el síndrome de secreción ectópica de ACTH tiene la mejor tasa de recuperación (80%; la mediana de tiempo de recuperación es siete meses), el Cushing suprarrenal tiene la tasa más baja (40%; la mediana de tiempo de recuperación es 30 meses) y la enfermedad de Cushing es intermedia (60%; con una mediana de tiempo de recuperación de 17 meses).
Exceso de mineralocorticoides primario
Epidemiología
Después de la primera descripción de un paciente con un adenoma suprarrenal productor de aldosterona (síndrome de Conn), se pensó que el exceso de mineralocorticoides representaba una causa rara de hipertensión. Sin embargo, en estudios que valoran de manera sistemática a todas las personas con hipertensión, ahora se ha identificado una prevalencia mucho mayor, que va de cinco a 12%. La prevalencia es mucho mayor cuando los pacientes son preseleccionados por hipertensión hipopotasémica o hipertensión resistente a fármacos.
Etiología
La causa más frecuente de exceso de mineralocorticoides primario (suprarrenal) es el aldosteronismo primario, lo que refleja el exceso de producción de aldosterona por la zona glomerulosa suprarrenal. De manera tradicional, los principales subtipos de la enfermedad son aldosteronismo primario bilateral por hiperplasia suprarrenal micronodular bilateral (la forma más común) y adenoma unilateral productor de aldosterona (síndrome de Conn) (cuadro 3). Aunque evidencias recientes han demostrado que el aldosteronismo primario existe en un espectro difuso entre los límites de las formas bilateral y unilateral, su distinción binaria respalda el manejo clínico, debido a que las formas unilaterales se favorecen de cirugía potencialmente curativa, mientras que la terapia con antagonistas de los receptores de mineralocorticoides es el tratamiento de elección para las formas bilaterales. Se han identificado mutaciones somáticas en conductos y enzimas responsables de incrementar el flujo de sodio y calcio al interior de las células de la zona glomerulosa suprarrenal, como causas prevalentes de adenomas suprarrenales productores de aldosterona (cuadro 3) y, en el caso de mutaciones de línea germinal, también de formas familiares de aldosteronismo primario. Sin embargo, la hiperplasia suprarrenal bilateral como causa de exceso de mineralocorticoides suele ser micronodular, pero también puede contener nódulos más grandes que podrían confundirse con un adenoma unilateral. Deben sospecharse las formas genéticas de aldosteronismo primario (que representan hasta 6% de los casos) en pacientes con hipertensión grave diagnosticada durante la niñez o la adultez temprana (cuadro 3). En circunstancias excepcionales, el hiperaldosteronismo primario es producido por un ACC. Deben sospecharse carcinomas en sujetos jóvenes y en personas con tumores grandes, porque los adenomas benignos productores de aldosterona por lo general miden < 2 cm de diámetro.
|
CAUSAS DEL EXCESO DE MINERALOCORTICOIDES PRIMARIO |
MECANISMO |
% |
|
Aldosteronismo primario esporádico |
|
94-99 |
|
Adenoma suprarrenal (síndrome de Conn) |
El exceso autónomo de aldosterona puede ser ocasionado por mutaciones somáticas (intratumorales) en el conducto del potasio GIRK4 (codificado por KCNJ5; identificado como causa de enfermedad en 40–70% de los adenomas productores de aldosterona y vinculado con sobreproducción de esteroides híbridos). Otras causas incluyen mutaciones somáticas que afectan a la subunidad α de la Na+/K+-ATPasa (codificada por ATP1A1), la ATPasa 3 de la membrana plasmática transportadora de calcio (codificada por ATP2B3), los conductos de calcio activados por voltaje CaV1.3 y CaV3.2 (codificados por CACNA1D y CACNA1H, en dicho orden) y el conducto de cloro ClC-2 (codificado por CLCN2). Todas las mutaciones incrementan la abundancia de CYP11B2 y por lo tanto la síntesis de aldosterona. De manera inusual, se han observado mutaciones somáticas que afectan a la β-catenina (codificada por CTNNB1), solas o combinadas con mutaciones en el gen CACNA1D y se presume que promueven un incremento en el número de células productoras de aldosterona. |
30-40 |
|
Hiperplasia suprarrenal bilateral |
Exceso de aldosterona autónomo bilateral por hiperplasia difusa productora de aldosterona y/o micronódulos múltiples productores de aldosterona. Se han descrito mutaciones somáticas en el gen CACNA1D en 58% de los micronódulos productores de aldosterona de pacientes con exceso de aldosterona bilateral sometidos a suprarrenalectomía como tratamiento no estandarizado. |
60-70 |
|
Aldosteronismo familiar primario |
|
1-6 |
|
Tipo 1 (también conocido como hiperaldosteronismo remediable con glucocorticoides o hiperaldosteronismo suprimible con dexametasona) |
Condición autosómica dominante. El entrecruzamiento entre los genes CYP11B1 y CYP11B2 genes provoca producción de aldosterona promovida por ACTH. Se caracteriza por hipertensión arterial grave en niños o adultos jóvenes (a menudo < 20 años de edad), con un alto riesgo de eventos cardiovasculares incluyendo ictus hemorrágico a edad temprana por rotura de aneurismas intracraneales. Los análisis de esteroidogénesis suprarrenal revelan sobreproducción de esteroides híbridos. |
|
|
Tipo 2 |
Condición autosómica dominante. Es la forma más común (1% a 6% de adultos con aldosteronismo primario). Se debe a mutaciones de línea germinal en el gen CLCN2. Presentación fenotípica variable, que por lo general incluye hipertensión arterial de inicio temprano. |
|
|
Tipo 3 |
Condición autosómica dominante. Mutaciones de línea germinal en el gen KCNJ5. Se caracteriza por hipertensión arterial de aparición temprana (< 20 años) con hiperplasia macronodular suprarrenal bilateral masiva. Los análisis de esteroidogénesis suprarrenal revelan sobreproducción de esteroides híbridos. |
|
|
Tipo 4 |
Condición autosómica dominante. Mutaciones de línea germinal en el gen CACNA1H. Se caracteriza por hipertensión arterial de aparición temprana (< 20 años), que en algunos casos puede vincularse con trastornos del desarrollo. |
|
|
Aldosteronismo primario, convulsiones y anormalidades neurológicas (PASNA) |
Mutaciones de novo en el gen CACNA1D. Se caracteriza por hipertensión arterial, convulsiones, anormalidades neurológicas, hiperinsulinismo congénito y anormalidades cardiacas que aparecen en la niñez. |
|
|
Otras causas (inusuales) |
|
<1 |
|
Síndrome de exceso aparente de mineralocorticoides (SAME, Syndrome of apparent mineralocorticoid excess) |
Mutaciones en el gen HSD11B2 restringen inactivación renal de cortisol a cortisona, lo que ocasiona una activación excesiva de MR por parte del cortisol (la inhibición de la deshidrogenasa de 11β-hidroxiesteroide tipo 2 por consumo excesivo de orozuz tiene efectos similares). |
|
|
Síndrome de Cushing |
El exceso de cortisol excede la capacidad de HSD11B2 para inactivar el cortisol y transformarlo en cortisona, lo que resulta en una sobreestimulación del receptor mineralocorticoide. |
|
|
Resistencia a glucocorticoides |
El aumento de la producción de cortisol por mutaciones en el receptor de glucocorticoides provoca una sobreestimulación del receptor mineralocorticoide por el cortisol. |
|
|
Carcinoma corticosuprarrenal |
Exceso autónomo de aldosterona y/o DOC. |
|
|
Hiperplasia suprarrenal congénita |
Acumulación de DOC a causa de mutaciones en los genes CYP11B1 o CYP17A1. |
|
|
Hipertensión arterial inducida por progesterona |
La progesterona actúa como un ligando anormal por mutaciones en el gen del MR. |
|
|
Síndrome de Liddle |
Las subunidades β o γ mutantes de ENaC disminuyen la degradación del ENaC, lo que mantiene el conducto de membrana en conformación abierta durante más tiempo, potenciando así la acción de los mineralocorticoides. |
|
Cuadro 3. Causas del exceso de mineralocorticoides primario. Abreviaturas: ACTH, hormona adrenocorticotrópica; DOC, desoxicorticosterona; ENaC, conducto de sodio epitelial; GR, receptor de glucocorticoides; HSD11B2, deshidrogenasa de 11β-hidroxiesteroide tipo 2; MR, receptor de mineralocorticoides.
Una causa poco frecuente de exceso de aldosterona es el aldosteronismo remediable con glucocorticoides (GRA, glucocorticoid-remediable aldosteronism), o aldosteronismo familiar primario tipo 1, que es causado por un gen quimérico resultado del entrecruzamiento de secuencias promotoras entre los genes CYP11B1 y CYP11B2 que participan en la síntesis de glucocorticoides y mineralocorticoides, en dicho orden (fig. 1). Este reordenamiento pone la transcripción de CYP11B2 bajo el control de la señalización del receptor de ACTH; por ello, la producción de aldosterona es regulada por ACTH en vez de por renina. Los antecedentes familiares pueden ser de utilidad, porque hay evidencia de transmisión dominante de hipertensión arterial. Es importante identificar este trastorno, porque puede estar vinculado con hipertensión arterial de inicio temprano y apoplejía. Además, la supresión de glucocorticoides puede reducir la producción de aldosterona. Las glándulas suprarrenales de pacientes con GRA producen niveles altos de esteroides híbridos 18-oxocortisol y 18-hidroxicortisol por la coexistencia de las actividades enzimáticas de CYP11B1 y CYP11B2 en las mismas células productoras de esteroides, que por lo general se segregan a las zonas fasciculada y glomerulosa, en dicho orden. De manera similar, niveles elevados de esteroides híbridos se observan en pacientes con adenomas productores de aldosterona que albergan mutaciones somáticas en el gen KCNJ5 (que codifica el conducto de potasio GIRK4) y en sujetos con aldosteronismo familiar primario por mutaciones de línea germinal en el gen KCNJ5 (cuadro 3). Los esteroides híbridos los producen las glándulas suprarrenales normales en concentraciones muy bajas y pueden, por lo tanto, medirse en la sangre y la orina para identificar estas condiciones.
En el cuadro 3 se enlistan otras causas inusuales de exceso de mineralocorticoides primario. Una causa importante es el exceso de unión y activación del receptor de mineralocorticoides por otros esteroides diferentes a la aldosterona. El cortisol actúa como un potente mineralocorticoide si escapa a la inactivación eficiente a cortisona por la 11β-HSD2 en el riñón (fig. 7). Esto puede ser ocasionado por mutaciones inactivadoras en el gen HSD11B2, lo que provoca un síndrome de exceso aparente de mineralocorticoides (SAME, syndrome of apparent mineralocorticoid excess), que de manera característica se manifiesta con hipertensión hipopotasémica grave en la infancia. Sin embargo, las mutaciones más leves pueden ocasionar hipertensión con niveles normales de potasio que se manifiesta en la etapa adulta (SAME tipo II). La inhibición de la 11β-HSD2 por consumo excesivo de orozuz también genera hipertensión hipopotasémica, como sucede cuando se sobrepasa la capacidad de transformación de la 11β-HSD2 por el exceso de cortisol en el síndrome de Cushing. La desoxicorticosterona (DOC, deoxycorticosterone) también se une y activa al receptor de mineralocorticoides y puede ocasionar hipertensión arterial si se incrementan sus concentraciones circulantes. Esto puede surgir por la secreción autónoma de DOC por un carcinoma corticosuprarrenal, pero también cuando la DOC se acumula como consecuencia de un bloqueo suprarrenal enzimático, como se observa en la hiperplasia suprarrenal congénita (CAH, congenital adrenal hyperplasia) por deficiencia de CYP11B1 (11β-hidroxilasa) o CYP17A1 (17α-hidroxilasa) (fig. 1). La progesterona puede ocasionar hipertensión hipopotasémica en personas excepcionales que portan mutaciones del receptor de mineralocorticoides que potencian la unión y la activación mediadas por la progesterona; de manera fisiológica, la progesterona de manera normal ejerce una actividad antimineralocorticoide. Por último, el exceso de actividad mineralocorticoide puede ser consecuencia de mutaciones en las subunidades β o γ del ENaC, lo que interrumpe su interacción con Nedd4 (fig. 7) y de esta manera disminuye la internalización y la degradación del receptor. El ENaC constitutivamente activo genera hipertensión hipopotasémica, que provoca un trastorno autosómico dominante llamado síndrome de Liddle.
Manifestaciones clínicas
La activación excesiva del receptor de mineralocorticoides ocasiona pérdida de potasio y mayor retención de sodio, lo cual expande los volúmenes extracelular y plasmático. El aumento de actividad del ENaC también ocasiona pérdida de hidrógeno, que puede generar alcalosis metabólica. La aldosterona también tiene efectos directos sobre el sistema vascular, donde aumenta la remodelación cardiaca y disminuye la distensibilidad. El exceso de aldosterona puede ocasionar daño directo al miocardio y a los glomérulos renales, además del daño secundario por hipertensión arterial sistémica.
El dato clínico distintivo del exceso de mineralocorticoides es la hipertensión hipopotasémica; sin embargo, solo 10% a 40% de los pacientes con aldosteronismo primario presenta hipopotasemia. El sodio sérico tiende a ser normal por la retención simultánea de líquido, que en algunos casos puede causar edema periférico. La hipomagnesiemia es también un hallazgo frecuente. La hipopotasemia puede exacerbarse con el tratamiento farmacológico con tiazidas, que incrementa el aporte de sodio a los túbulos renales distales, conduciendo así la excreción de potasio. La hipopotasemia grave puede vincularse con poliuria, intolerancia a la glucosa, debilidad muscular, miopatía proximal manifiesta e incluso arritmias, rabdomiólisis y parálisis hipopotasémica. La alcalosis grave favorece la presencia de calambres musculares y en casos graves puede ocasionar tetania.
Es importante señalar que los pacientes con aldosteronismo primario presentan tasas más altas de osteoporosis, diabetes tipo 2 y disfunción cognitiva. Una proporción significativa de individuos con aldosteronismo primario sufre secreción de cortisol autónoma leve (MACS, mild autonomous cortisol secretion) concurrente, un cuadro que también se denomina síndrome de Connshing.
Diagnóstico
La evaluación diagnostica para el exceso de mineralocorticoides no se recomienda en la actualidad para todos los pacientes con hipertensión arterial, sino que debe restringirse a sujetos que exhiben hipertensión vinculada con resistencia a fármacos, hipopotasemia, una tumoración suprarrenal, inicio de enfermedad antes de los 40 años de edad, o antecedentes familiares de aldosteronismo primario (fig. 12). La prueba de detección aceptada comprende la medición concurrente de renina y aldosterona plasmáticas más el cálculo subsecuente de la proporción aldosterona-renina (ARR, aldosterone-renin ratio) (fig. 12); es necesario normalizar los resultados de potasio sérico antes de la prueba. Interrumpir la medicación antihipertensiva puede ser difícil, sobre todo en pacientes con hipertensión arterial grave. Por lo tanto, con fines prácticos, en primera instancia, el individuo puede continuar con sus medicaciones antihipertensivas habituales, excepto que los antagonistas de los receptores de mineralocorticoides deben interrumpirse al menos cuatro a seis semanas antes de cuantificar la ARR. El resto de los fármacos antihipertensivos también puede afectar el resultado de la prueba de la ARR (p. ej., el tratamiento con β-bloqueadores puede ocasionar resultados positivos falsos y los inhibidores de la ACE/AT1R pueden causar resultados negativos falsos en casos más leves y puede ser necesario descontinuarlos por dos a cuatro semanas en caso de que se requiera repetir las pruebas) (cuadro 4). La decisión de depurar fármacos con potencial de interferir tiene que individualizarse.
|
EFECTO SOBRE LA ARR |
FÁRMACOS ANTIHIPERTENSORES |
OTROS FÁRMACOS |
OTRAS CONDICIONES |
|
Posibles resultados negativos falsos (por incremento de renina y/o aldosterona) |
Antagonistas de los MR; diuréticos; antagonistas de DHP-calcio; inhibidores de la ACE; antagonistas de los AT1R; aliskiréna |
Inhibidores de los SGLT2; SSRI |
Hipopotasemia; restricción dietética de sal; hipertensión arterial maligna; hipertensión renovascular; embarazo |
|
Posibles resultados positivos falsos (por reducción de renina y/o aldosterona) |
β-bloqueadores; clonidina; α-metildopa; aliskiréna |
AINE; anticonceptivos orales con estrógenos |
Deterioro de la función renal con hiperpotasemia; fase lútea del ciclo menstrual en mujeres premenopáusicas |
|
Efecto insignificante |
Antagonistas del calcio no dihidropiridínicos; antagonistas de los receptores α1-adrenérgicos; hidralazina |
/ |
/ |
Cuadro 4. Efectos de medicaciones y otras condiciones sobre la proporción aldosterona-renina (ARR). aEl aliskirén, un inhibidor directo de la renina, puede provocar resultados negativos falsos si se mide la concentración de renina directa, puesto que provoca reducción de la actividad de la renina plasmática.
Abreviaturas: ACE, enzima convertidora de angiotensina; AT1R, receptor de angiotensina II tipo 1; MR, receptor de mineralocorticoides; NSAID, fármacos antiinflamatorios no esteroideos; SGLT2, cotransportador 2 de sodio-glucosa; SSRI, inhibidor selectivo de la recaptación de serotonina.
Las guías internacionales actuales sugieren que un resultado positivo de vigilancia de la ARR debe ser > 750 pmol/L por ng/ml por hora, con una concentración de aldosterona concurrentemente normal alta o incrementada (fig. 12). Sin embargo, en la práctica clínica se utilizan de forma rutinaria límites de ARR más bajos, puesto que se reconoce cada vez más que de lo contrario podrían omitirse formas más leves de aldosteronismo primario. Otra consideración es que si solo se contempla la ARR, la probabilidad de obtener un valor positivo falso incrementa cuando las concentraciones de renina son muy bajas. Las características de los ensayos bioquímicos también son importantes. Algunos laboratorios miden la actividad de la renina plasmática, en tanto que otros cuantifican las concentraciones de renina plasmática. Los ensayos basados en anticuerpos para medir aldosterona sérica carecen de la confiabilidad de la espectrometría de masas en tándem, pero esta no se encuentra disponible de forma ubicua.
La confirmación diagnóstica de exceso de mineralocorticoides en un paciente con resultado positivo de la ARR debe realizarla un endocrinólogo, puesto que el ensayo carece de validación optimizada. La evaluación más directa es la prueba de infusión salina, que consiste en administrar por vía IV dos litros de solución salina fisiológica en un periodo de 4 h. Si la aldosterona no se suprime a < 170 pmol/L (6 ng/100 ml) en sedestación o < 140 pmol/L (5 ng/dl en decúbito, esto indica exceso autónomo de mineralocorticoides. Pruebas alternativas son el examen de carga de sodio oral (300 mmol NaCl/día durante tres días) o la prueba de supresión con fludrocortisona (0.1 mg cada 6 h con 30 mmol de NaCl cada 8 h durante cuatro días); la última de ellas puede ser difícil por el riesgo de hipopotasemia grave y aumento de la hipertensión. En individuos con hipertensión hipopotasémica evidente, ARR muy positiva y niveles concurrentemente incrementados de aldosterona, no es necesario realizar pruebas confirmatorias.
Diagnóstico diferencial y tratamiento
Tras establecer el diagnóstico de hiperaldosteronismo, lo siguiente es utilizar las imágenes de las glándulas suprarrenales para valorar la causa. El método de elección es la CT de cortes finos de la región suprarrenal, porque ofrece una excelente visualización de la morfología suprarrenal y la mayor parte de los adenomas productores de aldosterona mide < 2 cm. La CT identifica con facilidad tumores grandes con sospecha de malignidad, pero puede pasar por alto lesiones < 5 mm. Solo es necesario diferenciar entre hiperplasia micronodular bilateral y un adenoma unilateral, si es factible y deseable una estrategia quirúrgica. En consecuencia, el muestreo selectivo de vena suprarrenal (AVS, adrenal vein sampling) solo debe realizarse en candidatos a cirugía sin lesión obvia en la CT o evidencia de una lesión unilateral pero menores de 35 años de edad, puesto que estos últimos tienen una probabilidad alta de albergar un adenoma suprarrenal endocrino inactivo coincidente (fig. 12). El AVS sirve para comparar las concentraciones de aldosterona en la vena cava inferior y entre las venas suprarrenales derecha e izquierda. El AVS exige la medición concurrente de cortisol para documentar la colocación correcta del catéter en las venas suprarrenales y debe demostrar un gradiente de cortisol > 2–3 en condiciones basales entre la vena cava y cada una de las venas suprarrenales. La lateralización se confirma por una proporción aldosterona/cortisol que es al menos dos veces mayor en un lado que en el otro en condiciones basales. El AVS es un procedimiento complejo que tiene que realizar un radiólogo intervencionista muy especializado. Aun así, puede ser difícil canalizar de manera correcta la vena suprarrenal derecha, lo cual, si no se logra, invalida el procedimiento. Tampoco hay acuerdo sobre si las dos venas suprarrenales deben cateterizarse de manera simultánea o sucesiva y si la estimulación con ACTH potencia el valor diagnóstico del AVS. En fecha reciente, la PET-CT con una forma etiquetada de metomidato (un análogo metilado del etomidato que se une a CYP11B1 y a CYP11B2) se validó como una alternativa no invasiva de AVS. La PET-CT con [11C] metomidato y pretratamiento con dexametasona para suprimir la actividad de CYP11B1 puede diferenciar entre formas unilaterales y bilaterales de aldosteronismo primario y tiene un desempeño similar al AVS para pronosticar éxito clínico y bioquímico después de la suprarrenalectomía.
Los sujetos mayores de 35 años con aldosteronismo primario confirmado, una lesión unilateral en los estudios de CT y sin sospecha clínica de formas familiares pueden ir directo a cirugía, la cual también está indicada en pacientes con lateralización confirmada, documentada por un procedimiento de AVS válido o una PET-CT con [11C] metomidato. La suprarrenalectomía laparoscópica es el abordaje preferido. Los pacientes que no son elegibles para cirugía o que tienen evidencia de hiperplasia bilateral basada en CT o en AVS, deben recibir tratamiento médico (fig. 12). El tratamiento médico, que también debe considerarse antes de la cirugía para evitar hipoaldosteronismo posoperatorio, consiste en primera instancia en la administración de espironolactona, un antagonista de los MR. Este fármaco puede iniciarse con 12.5 a 50 mg dos veces al día y ajustarse a un máximo de 400 mg al día para controlar la presión sanguínea y normalizar el potasio. Sus efectos secundarios consisten en irregularidades menstruales, disminución de la libido y ginecomastia. También se puede utilizar eplerenona (un antagonista más selectivo de los receptores de mineralocorticoides). Las dosis inician con 25 mg cada 12 h, y pueden dosificarse hasta 200 mg al día. Otro fármaco útil es la amilorida, un diurético ahorrador de potasio (5 a 10 mg cada 12 h).
En pacientes con morfología suprarrenal normal y antecedentes familiares de hipertensión grave de inicio temprano, el diagnóstico de GRA debe tenerse en cuenta y puede evaluarse mediante pruebas genéticas. El tratamiento de GRA consiste en la administración de dexametasona; se utiliza la dosis más baja posible para controlar la presión arterial. Algunos individuos también necesitan tratamiento adicional con un antagonista de los MR.
El diagnóstico de exceso de mineralocorticoides no vinculado con aldosterona se basa en la documentación de renina y aldosterona suprimidas en presencia de hipertensión hipopotasémica. La mejor manera de llevar a cabo esta prueba es utilizando el perfil de metabolitos de esteroides urinarios mediante cromatografía de gases/espectrometría de masas (GC, gas chromatography/MS, mass spectrometry). El aumento de la proporción cortisol libre/cortisona libre sugiere SAME y puede tratarse con dexametasona. El perfil de esteroides por GC/MS también detecta los esteroides vinculados a deficiencia de CYP11B1 y CYP17A1 o el patrón irregular de secreción de esteroides en un carcinoma corticosuprarrenal productor de DOC (fig. 12). Si el perfil de la GC/MS es normal, debe pensarse en el síndrome de Liddle. Es muy sensible al tratamiento con amilorida pero no responderá a la terapia con antagonistas de los MR, porque el defecto se debe a un ENaC constitutivamente activo.
Estudio del paciente: Tumoración suprarrenal descubierta accidentalmente
Epidemiología
Las tumoraciones suprarrenales descubiertas de forma incidental Son frecuentes y en general se les denomina “incidentalomas” suprarrenales. Tienen una prevalencia de al menos 2–5% en la población general cuando se documentan por CT y series de necropsias. La prevalencia aumenta con la edad; alrededor de 1% de personas a los 40 años de edad y 7% a los 70 años de edad presentan una tumoración suprarrenal. El uso generalizado de estudios de imagen con cortes transversales ha incrementado la prevalencia reconocida.
Etiología
La mayor parte de los tumores suprarrenales solitarios comprende neoplasias monoclonales. Varios síndromes genéticos, incluyendo MEN-1 (MEN1), MEN-2 (RET), complejo de Carney (PRKAR1A), McCune-Albright (GNAS1), Li Fraumeni (TP53), Lynch (MLH1, MSH2, MSH6, PMS2, EPCAM) y poliposis adenomatosa familiar (APC, familial adenomatous polyposis), pueden tener tumores corticosuprarrenales como una de sus características. Por otra parte, la enfermedad de Von-Hippel-Lindau (VHL), el MEN 2, el síndrome de paraganglioma tipos 1/4/5 (SDHD/SDHB/SDHA), la neurofibromatosis tipo 1 (NF1) y la leiomiomatosis hereditaria y cáncer de células renales (FH) se encuentran entre los síndromes genéticos vinculados con feocromocitomas.
Hasta 50% de los nódulos suprarrenales es activo en términos hormonales, por un adenoma corticosuprarrenal productor de cortisol o aldosterona o por un feocromocitoma vinculado con exceso de catecolaminas (cuadro 5). El ACC es poco frecuente, pero es la causa de una masa suprarrenal en 4% de los pacientes. Sin embargo, las metástasis que provienen de otro tumor de tejido sólido son una causa adicional de incidentalomas suprarrenales y representan hasta 40% de las masas suprarrenales en pacientes sometidos a estudios de imagen para estadificación de tumores o monitorización de seguimiento (cuadro 5).
|
TUMORACIÓN |
PREVALENCIA APROXIMADA (%) |
|
Benigna |
|
|
Adenoma corticosuprarrenal |
|
|
Endocrino-inactivo |
40-70 |
|
Productor de cortisol (secreción de cortisol autónoma leve) |
20-50 |
|
Productor de aldosterona |
2-5 |
|
Productor de cortisol (síndrome de Cushing evidente) |
1-4 |
|
Feocromocitoma |
1-5 |
|
Mielolipoma suprarrenal |
3-6 |
|
Ganglioneuroma suprarrenal |
1 |
|
Quiste y pseudoquiste suprarrenales |
1 |
|
Hematoma suprarrenal/infarto hemorrágico |
<1 |
|
Hemangioma suprarrenal |
<0.1 |
|
Indeterminada |
|
|
Oncocitoma corticosuprarrenal |
<1 |
|
Maligna |
|
|
Metástasis (con mayor frecuencia: mamas y pulmones) |
3-7 |
|
Carcinoma corticosuprarrenal |
0.4-4 |
|
Feocromocitoma maligno |
<1 |
|
Neuroblastoma suprarrenal |
<0.1 |
|
Linfomas (incluye linfoma suprarrenal primario) |
<0.1 |
Cuadro. Clasificación de las tumoraciones suprarrenales unilaterales. Nota: Agrandamiento suprarrenal bilateral/los tumores pueden ser causados por hiperplasia suprarrenal congénita, hiperplasia macronodular bilateral, hemorragia bilateral (síndrome antifosfolipídico o síndrome de Waterhouse-Friderichsen vinculado con septicemia), granuloma, amiloidosis o enfermedad infiltrativa, incluyendo tuberculosis.
Diagnóstico diferencial y tratamiento
Los pacientes con una tumoración suprarrenal > 1 cm necesitan una valoración diagnóstica. Es necesario responder dos preguntas clave: 1) ¿Secreta el tumor hormonas de manera autónoma que pudieran tener un efecto perjudicial en la salud? y 2) ¿La tumoración suprarrenal es benigna o maligna?
La secreción de hormonas por una masa suprarrenal se presenta a lo largo de un proceso continuo, con un aumento gradual de las manifestaciones clínicas que es paralelo a las concentraciones hormonales. La exclusión del exceso de catecolaminas proveniente de un feocromocitoma que se origina en la médula suprarrenal es una parte mandatoria de la pruebas diagnósticas de las masas con lípidos escasos (fig. 13). En caso de hipertensión, es necesario descartar aldosteronismo primario. La producción excesiva de precursores de andrógenos suprarrenales, DHEA y su sulfato, es inusual y se observa con mayor frecuencia en el contexto de ACC, igual que los niveles aumentados de precursores de esteroides, como 17OHP. El exceso de cortisol es la anormalidad hormonal más observada en presencia de masas suprarrenales, y oscila entre síndrome de Cushing inusual con manifestaciones clínicas y MACS mucho más prevalente (cortisol al inicio de la mañana > 50 nmol/L [1.8 mcg/dl] después de la administración de 1 mg de dexametasona durante la noche en ausencia de características clínicas de Cushing). Los pacientes con MACS pueden presentar uno o más de los componentes del síndrome metabólico (p. ej., obesidad, diabetes tipo 2, hipertensión y dislipidemia), osteoporosis, riesgo incrementado de eventos cardiovasculares, debilidad, deterioro de la calidad de vida y mayor riesgo de mortalidad. El tratamiento óptimo de estos pacientes sigue siendo motivo de debate, e incluye suprarrenalectomía o tratamiento conservador con vigilancia a largo plazo y tratamiento de comorbilidades potencialmente atribuibles al exceso de cortisol.
Para diferenciar las masas suprarrenales benignas de las malignas los estudios de imagen son sensibles en términos relativos, aunque la especificidad es subóptima. La CT sin contraste es el procedimiento preferido para estudios de imagen de las glándulas suprarrenales (fig. 11). Los diagnósticos de carcinoma corticosuprarrenal, feocromocitoma y mielolipoma suprarrenal benigno son más probables conforme incrementa el diámetro de la masa suprarrenal. Sin embargo, el tamaño por sí solo es de poca utilidad predictiva; solo 80% de especificidad para diferenciar las tumoraciones benignas de las malignas cuando se utiliza un límite de 4 cm. Las metástasis son poco frecuentes, pero se observan con la misma frecuencia en masas suprarrenales de todos los tamaños. El valor de atenuación tumoral en CT no contrastadas tiene gran valor diagnóstico, puesto que muchos adenomas corticosuprarrenales tienen abundantes lípidos y, por lo tanto, presentan valores de atenuación bajos (p. ej., densidades < 20 unidades Hounsfield [HU, Hounsfield Units]). Sin embargo, un número similar de adenomas corticosuprarrenales tiene pocos lípidos y se presentan con HU mayores, lo que hace difícil diferenciarlos de los ACC, así como de los feocromocitomas, ambos de los cuales tienen de manera invariable valores altos de atenuación (densidades > 20 HU en escaneos antes de la administración de medio de contraste). En general, las lesiones benignas son redondeadas y homogéneas, en tanto que muchas lesiones malignas son lobuladas y heterogéneas. El feocromocitoma y el mielolipoma suprarrenal también pueden presentar características de lobulación y heterogeneidad. La MRI también permite visualizar las glándulas suprarrenales, pero con menor resolución que la CT. Sin embargo, debido a que no involucra exposición a radiación ionizante, la MRI se prefiere en niños, adultos jóvenes y durante el embarazo. Este estudio tiene gran utilidad en la caracterización de lesiones suprarrenales indeterminadas mediante análisis de desplazamiento químico; los tumores malignos muy pocas veces muestran pérdida de la señal en las MRI de fase opuesta; sin embargo, esto también puede observarse en una proporción de adenomas corticosuprarrenales benignos y los resultados de los análisis de desplazamiento químico por lo general son similares a los que se obtienen midiendo atenuación tumoral o CT sin contraste. La base de evidencias que sustentaba la utilidad de la PET con fluorodesoxiglucosa (FDG) al inicio era escasa, pero en la actualidad se está expandiendo; datos recientes indican que la falta de captación de FDG indica de manera confiable una masa corticosuprarrenal benigna. Sin embargo, debe tenerse cuidado en pacientes con antecedentes de cáncer extra-suprarrenal, puesto que las metástasis suprarrenales que surgen de estos cánceres en ocasiones no capturan FDG, como se observa de forma típica en el carcinoma de células renales. El carcinoma corticosuprarrenal de manera invariable es ávido por FDG; no obstante, su especificad es precaria y muchos adenomas suprarrenales benignos también capturan FDG.
En casos de tumoración suprarrenal, casi nunca se indica la aspiración con aguja fina (FNA, fine-needle aspiration) o biopsia guiada por CT. La FNA de un feocromocitoma puede ocasionar crisis hipertensivas que ponen en peligro la vida. La FNA de un carcinoma corticosuprarrenal lesiona la cápsula del tumor y puede generar metástasis en el trayecto de la aguja. Debe considerarse la FNA solo en pacientes con antecedentes de tumores malignos que no sean de origen suprarrenal y una tumoración suprarrenal recién identificada, después de la exclusión cuidadosa de un feocromocitoma, y si el resultado influirá sobre el tratamiento. Es importante destacar que en 25% de los pacientes con antecedentes de tumores malignos de origen no suprarrenal, una masa recién detectada mediante CT no es una metástasis. Si bien la FNA puede diagnosticar neoplasias malignas extrasuprarrenales, su capacidad para diferenciar entre lesiones corticosuprarrenales benignas y malignas es limitada y, por lo tanto, no debe utilizarse para diagnosticar ACC.
Las tumoraciones suprarrenales vinculadas con exceso hormonal confirmado o sospecha de cáncer por lo general se tratan con cirugía (fig. 13) o, si la suprarrenalectomía no es posible o deseable, con fármacos. La exclusión preoperatoria del exceso de glucocorticoides es muy importante para pronosticar la supresión posquirúrgica de la glándula suprarrenal contralateral, que exige la restitución de glucocorticoides alrededor de la cirugía y después de ella. Las tumoraciones suprarrenales de cualquier tamaño con una bioquímica endócrina normal en el momento del diagnóstico y un valor de atenuación < 10 HU en la CT sin medio de contraste, se consideran benignas (índice de malignidad < 0.5%) y no es necesario estudiarlas más (fig. 13). Esto aplica de manera similar a las tumoraciones suprarrenales < 4 cm de diámetro y valores de atenuación de 10 a 20 HU. En dichas tumoraciones < 4 cm y > 20 HU de atenuación y en aquellas > 4 cm y 10 a 20 HU, las probabilidades de malignidad siguen siendo muy bajas (< 5%) y pueden mitigarse ordenando estudios de imagen en intervalos (fig. 13). La mayor parte del riesgo de malignidad aplica a tumoraciones suprarrenales > 4 cm con una atenuación > 20 HU y estas masas requieren atención inmediata (fig. 13); tienen un riesgo de 50% de malignidad subyacente. El riesgo de carcinoma corticosuprarrenal es más alto en pacientes jóvenes (< 40 años), mientras que las metástasis de condiciones primarias extra-suprarrenales ocurren a todas las edades. En las masas suprarrenales que generan hallazgos sospechosos en los estudios de imagen (> 4 cm y > 20 HU), es posible hacer cirugía o tomar una biopsia; sin embargo, las últimas culminarán en la cirugía innecesaria de muchos tumores benignos. Existe una prueba diagnóstica reciente, la metabolómica de los esteroides urinarios, que tiene un valor predictivo positivo dos veces mayor que los estudios de imagen para detectar carcinoma corticosuprarrenal, que se basa en un “patrón específico maligno de esteroides” distintivo que se caracteriza por la presencia de metabolitos precursores de esteroides acumulados en orina de 24 horas.
Carcinoma corticosuprarrenal
El carcinoma corticosuprarrenal (ACC, adrenocortical carcinoma) es un cáncer poco frecuente con una incidencia anual de 1–2 casos por millón de habitantes en la población. El ACC en general se considera un tumor de gran malignidad; sin embargo, se presenta con una amplia variabilidad de características biológicas y comportamiento clínico. Las mutaciones somáticas del gen oncosupresor TP53 se observan en 25% de los ACC al parecer esporádicos. Las mutaciones de TP53 en la línea germinal causan el síndrome de Li-Fraumeni que se vincula con múltiples cánceres de órganos sólidos, incluido el ACC, y se identifican en 25% de los casos pediátricos de ACC; en Brasil, la mutación R337H en TP53 se encuentra en casi todos los pacientes pediátricos con ACC. Otros cambios genéticos identificados en ACC incluyen mutaciones de la línea germinal en genes que afectan la reparación de apareamientos erróneos en el DNA (síndrome de Lynch), como PRKAR1A (complejo de Carney), MEN1 y APC (poliposis adenomatosa familiar), y alteraciones somáticas en la ruta Wnt/catenina β y en el grupo del factor de crecimiento similar a insulina 2 (IGF2, insulin-like growth factor 2). En 90% de los carcinomas corticosuprarrenales se encuentra sobreexpresión de IGF2.
Los pacientes con tumores suprarrenales grandes sospechosos de cáncer deben ser atendidos por un equipo multidisciplinario especializado, formado por un endocrinólogo, un oncólogo, un cirujano, un radiólogo y un histopatólogo. La FNA no se indica en casos de sospecha de ACC: en primer lugar, la citología y también el estudio histopatológico de una biopsia del centro no pueden diferenciar las masas suprarrenales primarias benignas de las malignas; en segundo lugar, la FNA rompe la cápsula del tumor y puede incluso causar metástasis en el canal de la aguja. Incluso cuando está disponible todo el tumor, la diferenciación histopatológica entre lesiones corticosuprarrenales benignas y malignas es un reto diagnóstico. La clasificación histológica más utilizada es la puntuación de Weiss, que contempla el grado nuclear alto; la tasa mitótica (> 5/campo de alta resolución [HPF, high-power field]); mitosis atípica; < 25% de células claras; estructura difusa; presencia de necrosis, invasión venosa e invasión de estructuras sinusoidales y de la cápsula del tumor. La presencia de tres o más elementos sugiere ACC. No obstante, la FNA constituye una opción razonable cuando se buscan metástasis de un tumor extrasuprarrenal primario o algún otro tumor suprarrenal, como un ganglioneuroma.
A pesar de que 60–70% de los ACC muestra evidencia bioquímica de sobreproducción de esteroides, esto no es evidente en términos clínicos en muchos pacientes, debido a la producción un tanto ineficiente de esteroides por parte de las células cancerígenas corticosuprarrenales. La producción excesiva de glucocorticoides y precursores de andrógenos suprarrenales es muy común e indica malignidad.
La estadificación del tumor al momento del diagnóstico de ACC (cuadro 6) tiene repercusiones importantes en el pronóstico y se necesita realizar un escaneo de tórax y abdomen para buscar invasión de órganos locales, linfadenopatía y metástasis. Puede ser difícil determinar un origen suprarrenal mediante CT axiales estándar si los tumores son grandes e invasivos, pero las reconstrucciones con CT y la MRI son más informativas (fig. 14), usando múltiples planos y diferentes secuencias. La invasión vascular y de órganos adyacentes establece el diagnóstico de cáncer. La tomografía por emisión de positrones con 18-fluoro-2-desoxi-D-glucosa (18-FDG-PET, 18-Fluoro-2-deoxy–D–glucose positron emission tomography) es muy sensible para la detección de cáncer y puede utilizarse para identificar metástasis pequeñas o recurrencia local que podría no ser evidente en CT (fig. 14). Sin embargo, la FDG-PET tiene una especificidad limitada y, por lo tanto, no puede utilizarse para diferenciar las lesiones suprarrenales benignas de las malignas. Las metástasis en casos de ACC ocurren con más frecuencia en el hígado y los pulmones.
|
ETAPA DE LA ENSAT |
ETAPA DE TNM |
DEFINICIONES DE TNM |
|
I |
T1, N0, M0 |
T1, tumor ≤ 5 cm |
|
|
|
N0, ausencia de ganglios linfáticos positivos |
|
|
|
M0, sin metástasis a distancia |
|
II |
T2, N0, M0 |
T2, tumor > 5 cm |
|
|
|
N0, ausencia de ganglios linfáticos positivos |
|
|
|
M0, sin metástasis a distancia |
|
III |
T1-T2, N1, M0 |
N1, ganglio(s) linfático(s) positivo(s) |
|
|
T3-T4, N0-N1, M0 |
M0, sin metástasis a distancia |
|
|
|
T3, infiltración tumoral en el tejido circundante |
|
|
|
T4, invasión tumoral en los órganos adyacentes o trombo tumoral venoso en la vena cava o en la vena renal |
|
IV |
T1-T4, N0-N1, M1 |
M1, presencia de metástasis a distancia |
Cuadro 6. Sistema de clasificación para estadificar carcinoma corticosuprarrenal. Abreviaturas: ENSAT, European Network for the Study of Adrenal Tumors; TNM, tumor, ganglios linfáticos, metástasis.
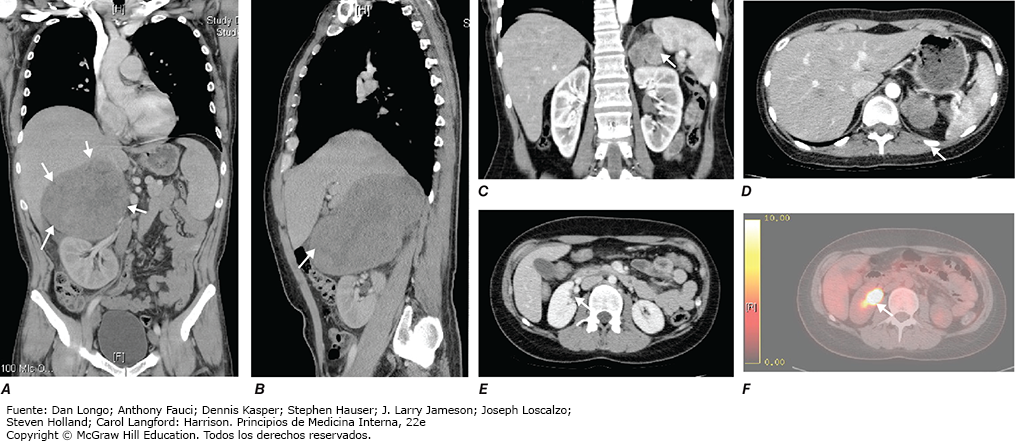
No existe un sistema de clasificación establecido para ACC, y la puntuación de Weiss no tiene valor pronóstico; el parámetro histopatológico pronosticador más importante es el índice de proliferación Ki67; un valor de Ki67 < 10% indica velocidad de crecimiento de lenta a moderada, en tanto que un valor de Ki67 ≥10% conlleva un mal pronóstico, que incluye riesgo alto de recurrencia y progresión rápida.
El ACC solo puede curarse si se detecta a tiempo y se remueve por completo con cirugía. La rotura de la cápsula durante la cirugía inicial, la presencia de metástasis en el momento del diagnóstico y el tratamiento inicial en un centro no especializado o por un cirujano no experto son factores fundamentales que determinan una supervivencia precaria. Si el tumor primario invade órganos adyacentes, debe considerarse la ablación en bloque del riñón y el bazo para disminuir el riesgo de recurrencia. Además, la disección de ganglios linfáticos regionales puede reducir aún más este riesgo. También es posible considerar la cirugía en un paciente con metástasis si hay exceso grave de hormonas vinculado con el tumor. Esta indicación necesita sopesarse con los riesgos quirúrgicos, que incluyen complicaciones tromboembólicas y el retraso subsiguiente de la aplicación de otras opciones terapéuticas. Los pacientes con ACC confirmado y extirpación exitosa del tumor primario deben recibir tratamiento adyuvante con mitotano (o,p′DDD), sobre todo los individuos con alto riesgo de recurrencia según las etapas III y IV de la European Network for the Study of Adrenal Tumors (ENSAT) o un índice de proliferación Ki67 ≥10%. El mitotano adyuvante debe continuarse durante al menos dos años, si se toleran los efectos colaterales. La vigilancia periódica de las concentraciones plasmáticas de mitotano es obligatoria (límites terapéuticos de 14 a 20 mg/L; las complicaciones neurotóxicas son más frecuentes con cifras > 20 mg/L). La dosis inicial de mitotano por lo general es de 500 mg tres veces al día. Después debe hacerse un aumento gradual en un lapso de días (saturación en dosis altas) o semanas (saturación en dosis bajas), conforme se tolere, hasta alcanzar una dosis máxima de 2000 mg tid. Una vez que se alcanzan concentraciones plasmáticas de mitotano dentro del rango terapéutico, puede ajustarse la dosis de mantenimiento entre 1000 y 1500 mg tres veces al día. El tratamiento con mitotano altera la síntesis de cortisol y, por lo tanto, se necesita restitución de glucocorticoides; esta requiere dosis de al menos el doble de las que se requieren de manera general en pacientes con insuficiencia suprarrenal (es decir, 40 a 60 mg al día, divididos en dos o tres tomas), puesto que el mitotano induce la actividad de la enzima CYP3A4 hepática, lo cual inactiva los glucocorticoides con rapidez. El mitotano también aumenta la globulina transportadora de cortisol circulante, con lo que disminuye la fracción disponible de cortisol libre. Las metástasis únicas pueden tratarse con cirugía o con ablación por radiofrecuencia, según sea apropiado. Si el tumor recurre o progresa durante el tratamiento con mitotano, se considera la quimioterapia citotóxica; el régimen de quimioterapia de primera línea establecido consiste en una combinación de cisplatino, etopósido y doxorrubicina, más la continuación del mitotano. Las metástasis óseas dolorosas responden a irradiación. La supervivencia global en casos de ACC aún es precaria; las tasas de supervivencia a cinco años son de 30 a 40%, y la mediana de supervivencia es de 15 meses en sujetos con ACC metastásico.
Insuficiencia suprarrenal
Epidemiología
La prevalencia de la insuficiencia suprarrenal permanente, que está bien documentada, es de cinco en 10 000 personas en la población general. El origen más frecuente de la enfermedad es hipotalámico-hipofisario, con una prevalencia de tres en 10 000, en tanto que la insuficiencia suprarrenal primaria tiene una prevalencia de dos en 10 000. Casi la mitad de los últimos casos es adquirida; la mayor parte es ocasionada por destrucción autoinmunitaria de las glándulas suprarrenales. La otra mitad es de origen genético y es ocasionada muy a menudo por distintos bloqueos enzimáticos en la esteroidogénesis suprarrenal que afectan la síntesis de glucocorticoides (hiperplasia suprarrenal congénita).
Es mucho más común la insuficiencia suprarrenal que se presenta por supresión del eje HPA como consecuencia del tratamiento con glucocorticoides exógenos, y se observa en 0.5% a 2% de la población en países desarrollados.
Etiología
La suprarrenalitis autoinmunitaria es la primera causa de insuficiencia suprarrenal primaria. La suprarrenalitis autoinmunitaria aislada causa 30 a 40% de los casos, en tanto que 60 a 70% presenta insuficiencia suprarrenal como parte de síndromes poliglandulares autoinmunitarios (APS, autoimmune polyglandular syndromes) (cuadro 7). La causa subyacente en 10% de los individuos afectados por APS es el síndrome poliglandular autoinmunitario tipo 1 (APS1), también denominado poliendocrinopatía autoinmunitaria con candidiasis y distrofia ectodérmica (APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy). El APS1 se transmite de forma autosómica recesiva y es ocasionado por mutaciones en el gen regulador autoinmunitario AIRE. Las condiciones autoinmunitarias vinculadas se superponen con las observadas en el síndrome poliglandular autoinmunitario tipo 2 (APS2); sin embargo, pueden incluir alopecia total, hipoparatiroidismo primario y, en casos poco comunes, linfoma. Los sujetos con APS1 presentan de manera invariable candidiasis mucocutánea crónica, que en general se manifiesta en la infancia y antecede a la insuficiencia suprarrenal por años o décadas. El APS2 es mucho más prevalente y se hereda de forma poligénica y se han confirmado sus vínculos con la región génica HLA-DR3 en el complejo mayor de histocompatibilidad y con regiones génicas distintas que participan en la regulación inmunológica (CTLA-4, PTPN22 y CLEC16A). La enfermedad autoinmunitaria coincidente incluye con mayor frecuencia enfermedad tiroidea autoinmunitaria, vitiligo e insuficiencia ovárica prematura. Otras características menos frecuentes son diabetes tipo 1 y anemia perniciosa ocasionada por deficiencia de vitamina B12.
|
DIAGNÓSTICO |
GEN |
CARACTERÍSTICAS VINCULADAS |
|
Síndrome poliglandular autoinmunitario tipo 1 (APS1) |
AIRE |
Hipoparatiroidismo, candidiasis mucocutánea crónica, otros trastornos autoinmunitarios y de manera inusual linfomas |
|
Síndrome poliglandular autoinmunitario tipo 2 (APS2) |
Vínculos con HLA-DR3, CTL-4 |
Hipotiroidismo, hipertiroidismo, insuficiencia ovárica prematura, vitiligo, diabetes mellitus tipo 1 y anemia perniciosa |
|
Suprarrenalitis autoinmunitaria aislada |
Vínculos con HLA-DR3, CTL4 |
Véase el cuadro 10 |
|
Hiperplasia suprarrenal congénita (CAH) |
CYP21A2, CYP11B1, CYP17A1, HSD3B2, POR |
DSD 46,XY e insuficiencia gonadal |
|
Hiperplasia suprarrenal congénita lipoidea (CLAH) |
STAR, CYP11A1 |
DSD 46,XY e insuficiencia gonadal |
|
Hipoplasia suprarrenal congénita (AHC) |
NR0B1 (DAX-1), NR5A1 (SF-1) |
Desmielinización del sistema nervioso central (ALD) o la médula espinal y los nervios periféricos (AMN) |
|
Suprarrenoleucodistrofia (ALD), suprarrenomieloneuropatía (AMN) |
ABCD1 |
Talla alta |
|
Deficiencia familiar de glucocorticoides |
MC2R |
Ninguna |
|
|
MRAP |
Ninguna |
|
|
STAR |
Ninguna |
|
|
NNT TXNRD2 |
Ninguna |
|
|
MCM4 |
Retraso del crecimiento y deficiencia de linfocitos citolíticos naturales |
|
Síndrome triple A |
AAAS |
Alácrima, acalasia y deterioro neurológico |
|
Síndrome de Smith-Lemli-Opitz |
SLOS |
Trastorno de la síntesis de colesterol vinculado con retraso mental, malformaciones craneofaciales y retraso del crecimiento |
|
Síndrome de Kearns-Sayre |
Deleciones de DNA mitocondrial |
Oftalmoplejía externa progresiva, degeneración retiniana pigmentaria, defectos de conducción cardiaca, insuficiencia gonadal, hipoparatiroidismo y diabetes mellitus tipo 1 |
|
Síndrome IMAGe |
CDKN1C |
Retraso del crecimiento intrauterino, displasia metafisaria y anomalías genitales |
|
Síndrome MIRAGE |
SAMD9 |
Mielodisplasia, infección, restricción del crecimiento, fenotipos genitales y enteropatía |
|
Deficiencia de liasa de esfingosina-1-fosfato |
SGPL1 |
Síndrome nefrótico resistente a esteroides, inmunodeficiencia, defectos neurológicos, ictiosis, hipotiroidismo primario y criptorquidia |
|
Infecciones suprarrenales |
|
Tuberculosis, VIH, CMV, criptococosis, histoplasmosis y coccidiodomicosis |
|
Infiltración suprarrenal |
|
Metástasis, linfomas, sarcoidosis, amiloidosis y hemocromatosis |
|
Hemorragia suprarrenal |
|
Septicemia meningocócica (síndrome de Waterhouse-Friderichsen) y síndrome antifosfolipídico primario |
|
Inducción por fármacos |
|
Mitotano, aminoglutetimida, abiraterona, trilostano, etomidato, ketoconazol, levoketoconazol, osilodrostat, suramina, mifepristona (RU486), interferón α, ribavirina, acetato de megestrol e inhibidores de los puntos de control inmunitarios (inusual) |
|
Suprarrenalectomía bilateral |
|
Por ejemplo, en el tratamiento del síndrome de Cushing o después de nefrectomía bilateral |
Cuadro 7. Causas de insuficiencia suprarrenal primaria. Abreviaturas: AIRE, regulador autoinmunitario; CMV, citomegalovirus; DSD, trastorno de desarrollo sexual; MC2R, receptor de ACTH; MCM4, homólogo 4 deficiente en el mantenimiento de minicromosomas; MRAP, proteína accesoria MC2R; NNT, transhidrogenasa de nucleótido de nicotinamida.
La suprarrenoleucodistrofia vinculada al cromosoma X tiene una incidencia de 1:20 000 varones y es consecuencia de mutaciones en el gen X-ALD, que codifica la proteína transportadora de la membrana peroxisómica ABCD1; su alteración da como resultado la acumulación de ácidos grasos de cadena muy larga (> 24 átomos de carbono). Cerca de 50% de los casos se manifiesta en la infancia temprana con enfermedad de la materia blanca que progresa con rapidez (suprarrenoleucodistrofia cerebral); 35% se presenta durante la adolescencia o en la vida adulta temprana con características neurológicas indicativas de afectación del sistema nervioso periférico y de la mielina (suprarrenomieloneuropatía [AMN, adrenomyeloneuropathy]); y en el 15% restante, la insuficiencia suprarrenal es la única manifestación patológica. Cabe destacar que las mutaciones distintas se manifiestan con penetrancia y fenotipos variables en las familias afectadas.
Otras causas poco frecuentes de insuficiencia suprarrenal son destrucción de las glándulas suprarrenales por infecciones, hemorragias e infiltración (cuadro 7); la suprarrenalitis tuberculosa aún es una causa frecuente en países en vías de desarrollo. Las metástasis suprarrenales con poca frecuencia ocasionan insuficiencia suprarrenal, y esto ocurre solo con metástasis bilaterales voluminosas.
Las causas congénitas de insuficiencia suprarrenal primaria, además de la CAH, son inusuales y ocasionan < 1% de los casos. Sin embargo, su elucidación ofrece información importante sobre el desarrollo y la fisiología suprarrenales. Las mutaciones que ocasionan insuficiencia suprarrenal primaria (cuadro 7) incluyen factores que regulan el desarrollo y la esteroidogénesis suprarrenal (DAX-1 y SF-1), la síntesis de colesterol, la importación y el desdoblamiento (DHCR7, StAR y CYP11A1), los elementos de la vía de respuesta suprarrenal a ACTH (MC2R y MRAP) (fig. 5) y los factores que participan en la regulación rédox (NNT y TXNRD2) y en la reparación del DNA (MCM4 y CDKN1C).
La insuficiencia suprarrenal secundaria (o central) es consecuencia de la disfunción del componente hipotalámico-hipofisario del eje HPA (cuadro 8). Si se excluye la supresión yatrogénica, la mayor parte de los casos se debe a tumores hipotalámicos o hipofisarios o a su tratamiento por cirugía o irradiación. Causas más inusuales son la apoplejía hipofisaria, ya sea como consecuencia de un adenoma hipofisario infartado o por la reducción transitoria de la irrigación sanguínea de la hipófisis durante cirugía o después de una hemorragia rápida vinculada al parto, también llamada síndrome de Sheehan. Es muy inusual que la deficiencia aislada de ACTH sea consecuencia de una enfermedad autoinmunitaria o infiltración hipofisaria (cuadro 8). Las mutaciones en POMC, precursora de ACTH, o en factores que regulan el desarrollo hipofisario son causas genéticas de deficiencia de ACTH (cuadro 8).
|
DIAGNÓSTICO |
GEN |
CARACTERÍSTICAS VINCULADAS |
|
Tumores hipofisarios (adenomas endocrinos activos e inactivos; carcinoma: muy inusual) |
|
Dependiendo del tamaño del tumor y su localización: alteración de los campos visuales (hemianopsia bilateral), hiperprolactinemia, hipotiroidismo secundario, hipogonadismo y deficiencia de hormona del crecimiento |
|
Otras lesiones por tumoraciones que afectan la región hipotalámica-hipofisaria |
|
Craneofaringioma, meningioma, ependimoma y metástasis |
|
Irradiación hipofisaria |
|
Radioterapia administrada para tumores hipofisarios, tumores cerebrales o irradiación craneoespinal en leucemia |
|
Hipofisitis autoinmunitaria |
|
Vinculada con frecuencia al embarazo; puede presentarse como panhipopituitarismo o como deficiencia de ACTH aislada; puede estar asociada a enfermedad tiroidea autoinmunitaria, con menor frecuencia vitiligo, insuficiencia ovárica prematura, diabetes mellitus tipo 1 y anemia perniciosa |
|
Apoplejía/hemorragia hipofisaria |
|
Infarto hemorrágico de adenomas hipofisarios grandes o infarto hipofisario secundario a hemorragia traumática grave (p. ej., cirugía o embarazo: síndrome de Sheehan) |
|
Infiltración hipofisaria |
|
Tuberculosis, actinomicosis, sarcoidosis, histiocitosis X, granulomatosis con poliangitis (Wegener) y metástasis |
|
Inducción por fármacos |
|
Exceso crónico de glucocorticoides (endógenos o exógenos), inhibidores de los puntos control inmunitarios, opioides, interferón α, ribavirina y acetato de megestrol |
|
Deficiencia de ACTH congénita aislada |
TBX19 (Tpit) |
|
|
Deficiencia combinada de hormonas hipofisarias (CPHD) |
PROP-1 |
Desarrollo progresivo de CPHD en el siguiente orden: GH, PRL, TSH, LH/FSH, ACTH |
|
|
HESX1 |
CPHD y displasia septo-óptica |
|
|
LHX3 |
CPHD y limitación para girar el cuello, e hipoacusia neurosensitiva |
|
|
LHX4 |
CPHD y anomalías cerebelosas |
|
|
SOX3 |
CPHD y retraso mental variable |
|
Deficiencia de proopiomelanocortina (POMC) |
POMC |
Obesidad de inicio temprano y pigmentación roja del cabello |
Cuadro 8. Causas de insuficiencia suprarrenal secundaria. Abreviaturas: ACTH, hormona adrenocorticotrópica; GH, hormona del crecimiento; LH/FSH, hormona luteinizante/hormona foliculoestimulante; PRL, prolactina; TSH, hormona estimulante de la tiroides.
Manifestaciones clínicas
En principio, las manifestaciones clínicas de la insuficiencia suprarrenal primaria (enfermedad de Addison) incluyen pérdida de la secreción de glucocorticoides y mineralocorticoides (cuadro 9). En la insuficiencia suprarrenal secundaria solo se observa deficiencia de glucocorticoides, puesto que la glándula suprarrenal se encuentra intacta y, por lo tanto, aún puede regularse mediante el sistema RAA. La secreción de andrógenos suprarrenales se altera tanto en la insuficiencia suprarrenal primaria como en la secundaria (cuadro 9). La enfermedad hipotalámica-hipofisaria puede ocasionar otras manifestaciones clínicas debido a la participación de otros ejes endocrinos (tiroides, gónadas, hormona del crecimiento, prolactina) o deterioro visual con hemianopsia bitemporal por compresión del quiasma óptico. Es importante reconocer que la insuficiencia suprarrenal yatrogénica ocasionada por supresión del eje HPA por glucocorticoides exógenos puede producir todos los síntomas vinculados con deficiencia de glucocorticoides (cuadro 9), si los glucocorticoides exógenos se interrumpen de manera súbita. Sin embargo, los pacientes tendrán una apariencia cushingoide por la exposición excesiva precedente a glucocorticoides.
|
Signos y síntomas ocasionados por deficiencia de glucocorticoides |
|
Fatiga y falta de energía |
|
Pérdida de peso y anorexia |
|
Mialgia y dolor articular |
|
Fiebre |
|
Anemia normocrómica, linfocitosis y eosinofilia |
|
TSH un poco aumentada (debido a pérdida de inhibición por retroalimentación de la liberación de TSH) |
|
Hipoglucemia (más frecuente en niños) |
|
Hipotensión arterial e hipotensión postural |
|
Hiponatriemia (por pérdida de inhibición por retroalimentación de la liberación de AVP) |
|
Signos y síntomas ocasionados por deficiencia de mineralocorticoides (solo en insuficiencia suprarrenal primaria) |
|
Dolor abdominal, náusea y vómito |
|
Mareo e hipotensión postural |
|
Deseo de consumir sal |
|
Hipotensión arterial e hipotensión postural |
|
Aumento de creatinina en suero (por pérdida de volumen) |
|
Hiponatriemia |
|
Hiperpotasemia |
|
Signos y síntomas ocasionados por deficiencia de andrógenos suprarrenales |
|
Falta de energía |
|
Piel seca y prurito (en mujeres) |
|
Pérdida de libido (en mujeres) |
|
Pérdida de vello púbico y axilar (en mujeres) |
|
Otros signos y síntomas |
|
Hiperpigmentación (solo en insuficiencia suprarrenal primaria) (por exceso de péptidos derivados de la proopiomelanocortina [POMC]) |
|
Piel pálida con color de alabastro (solo en insuficiencia suprarrenal secundaria) (por deficiencia de péptidos derivados de la POMC) |
Cuadro 9. Signos y síntomas de la insuficiencia suprarrenal. Abreviaturas: AVP, arginina vasopresina; TSH, hormona estimulante de la tiroides.
La insuficiencia suprarrenal crónica se manifiesta con signos y síntomas inespecíficos en términos relativos, como fatiga y falta de energía, que suelen retrasar o hacer que se pase por alto el diagnóstico (p. ej., como si fuera depresión o anorexia). Una característica distintiva de la insuficiencia suprarrenal primaria es la hiperpigmentación, que es ocasionada por un exceso de estimulación de ACTH sobre los melanocitos. La hiperpigmentación es más pronunciada en áreas cutáneas expuestas a mayor fricción o estrés tensional, y aumenta con la exposición a la radiación solar (fig. 15). Por el contrario, en la insuficiencia suprarrenal secundaria, la piel tiene una palidez similar al alabastro por la falta de secreción de ACTH.
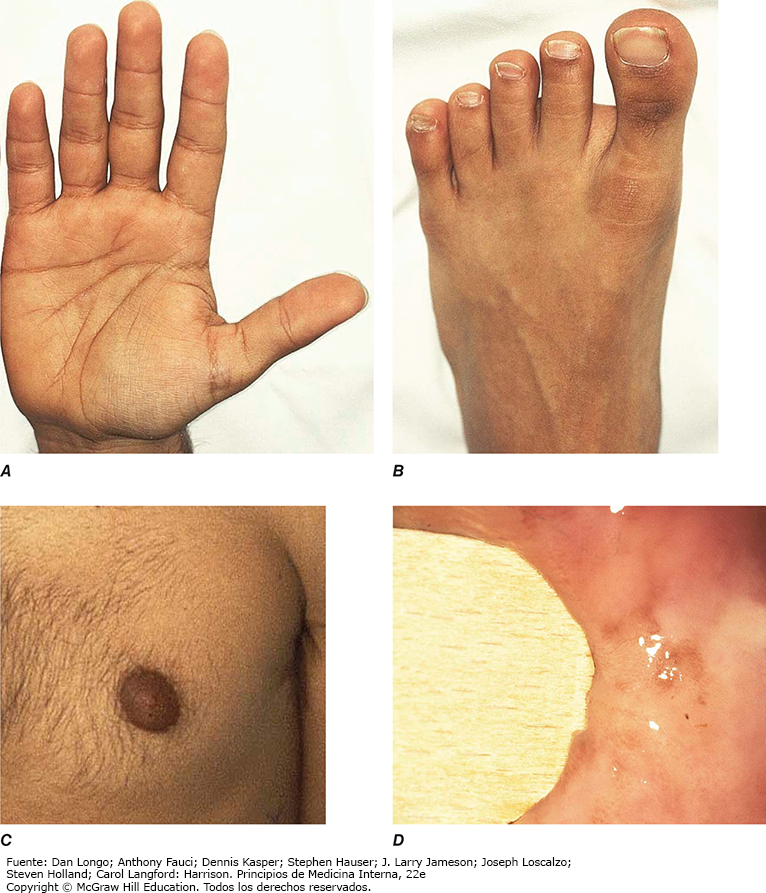
La hiponatriemia es una característica bioquímica de la insuficiencia suprarrenal primaria y se identifica hasta en 80% de los pacientes en el momento de la presentación. La hiperpotasemia ocurre en 40% de los individuos al momento del diagnóstico inicial. La hiponatriemia se debe sobre todo a deficiencia de mineralocorticoides, pero también puede ocurrir en la insuficiencia suprarrenal secundaria por disminución de la inhibición de la hormona antidiurética (ADH, antidiuretic hormone) liberada por el cortisol, lo que resulta en un síndrome leve de secreción inapropiada de hormona antidiurética (SIADH, syndrome of inappropriate secretion of antidiuretic hormone). La deficiencia de glucocorticoides también ocasiona un aumento leve de las concentraciones de TSH, que se normaliza en días a semanas después del inicio de la restitución de glucocorticoides.
La insuficiencia suprarrenal aguda, también llamada crisis suprarrenal, suele presentarse después de un periodo prolongado de molestias inespecíficas y se observa con más frecuencia en pacientes con insuficiencia suprarrenal primaria, por pérdida de la secreción de glucocorticoides y mineralocorticoides. La hipotensión ortostática puede progresar a choque hipovolémico. La insuficiencia suprarrenal puede imitar características de abdomen agudo, como dolor abdominal, náusea, vómito y fiebre. En algunos casos, la presentación inicial puede asemejarse una enfermedad neurológica, con disminución de la respuesta que progresa a estupor y coma. Una crisis suprarrenal puede iniciarse por una enfermedad concomitante, una cirugía u otro tipo de estrés, o por inactivación incrementada de glucocorticoides (p. ej., hipertiroidismo). Los datos prospectivos indican 8.3 crisis suprarrenales y 0.5 muertes relacionadas con crisis suprarrenales por cada 100 años-paciente.
Diagnóstico
Se establece con la prueba corta de cosintropina, un instrumento seguro y confiable con excelente valor diagnóstico predictivo (fig. 16). El límite para determinar insuficiencia se define por lo general con concentraciones de cortisol < 450 a 500 nmol/L (16 a 18 mcg/100 ml) con muestras obtenidas 30 a 60 min después de la estimulación con ACTH; el umbral exacto depende del ensayo disponible en la localidad; los límites por lo general son menores para ensayos basados en espectrometría de masa. Durante la fase inicial de la alteración del HPA (p. ej., durante las cuatro semanas de la insuficiencia hipofisaria), los pacientes aún pueden responder a la estimulación exógena con ACTH. En esta circunstancia, la prueba de tolerancia a la insulina es otra opción, pero es más invasiva y debe realizarse solo bajo vigilancia especializada (véase antes). La inducción de hipoglucemia está contraindicada en individuos con diabetes mellitus, enfermedad cardiovascular o antecedentes de crisis convulsivas. Las mediciones al azar de cortisol en suero tienen poca utilidad diagnóstica, porque las concentraciones basales de cortisol pueden estar bajas de manera coincidente por el ritmo diurno fisiológico de la secreción de cortisol (fig. 3). De manera similar, muchos pacientes con insuficiencia suprarrenal secundaria tienen concentraciones basales de cortisol normales en términos relativos, pero no desencadenan una respuesta de cortisol apropiada a la ACTH, lo cual solo puede evidenciarse mediante pruebas de estimulación. Es importante señalar que las pruebas para establecer el diagnóstico de insuficiencia suprarrenal no deben retrasar el tratamiento. Por lo tanto, en un sujeto con sospecha de crisis suprarrenal, es razonable obtener los niveles de cortisol basales, administrar tratamiento de restitución y posponer las pruebas formales de estimulación.
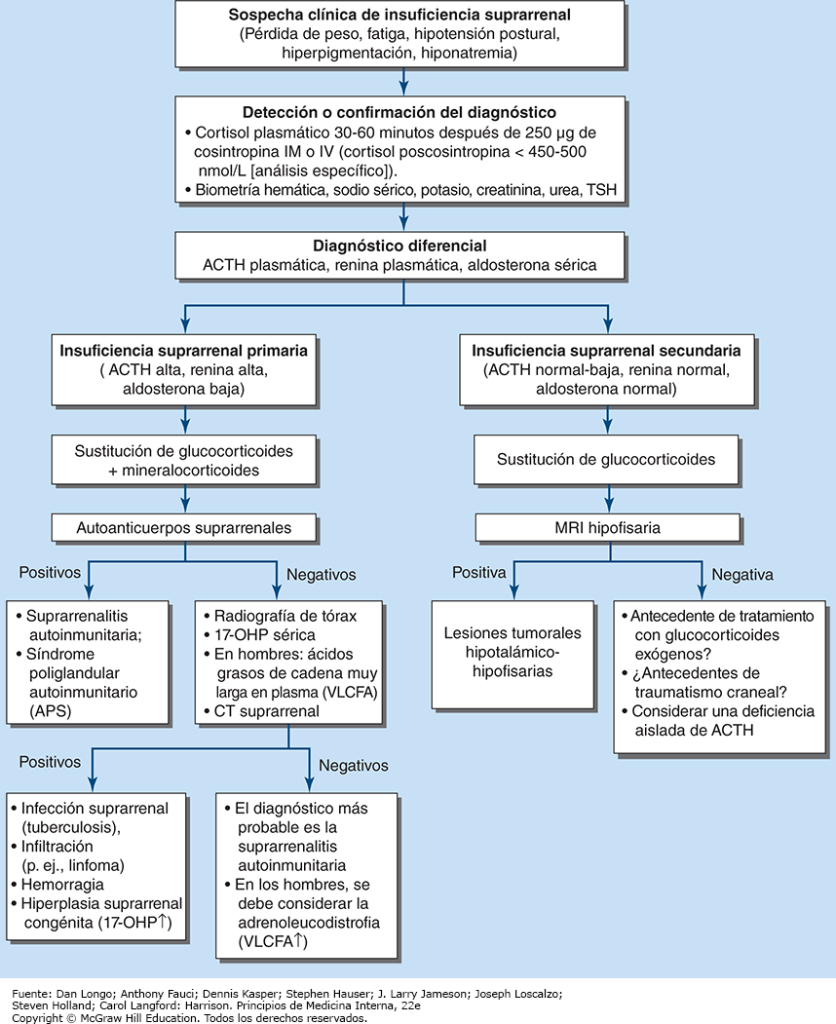
Después de confirmar el diagnóstico de insuficiencia suprarrenal, el siguiente paso es medir la ACTH en el plasma; concentraciones altas o demasiado bajas definen los orígenes primario y secundario de la enfermedad, en dicho orden (fig. 16). La renina aumentada en el plasma confirma la presencia de deficiencia de mineralocorticoides en la insuficiencia suprarrenal primaria. En la presentación inicial, los individuos con insuficiencia suprarrenal primaria deben someterse a revisión de autoanticuerpos contra esteroides como un marcador de suprarrenalitis autoinmunitaria. Si estas pruebas son negativas, se debe hacer una CT de las glándulas suprarrenales para descartar posibles hemorragias, infiltraciones o masas. En pacientes varones con autoanticuerpos negativos en plasma, se deben cuantificar los ácidos grasos de cadena muy larga para excluir X-ALD. A los sujetos con concentraciones demasiado bajas de ACTH, con deficiencia de cortisol confirmada, se les tiene que realizar una MRI de hipotálamo y la hipófisis. Los hallazgos que sugieren apoplejía hipofisaria previa, como cefalea grave de inicio súbito o antecedentes de traumatismos craneoencefálicos, deben investigarse con cuidado, en particular en personas sin lesiones evidentes en estudios de MRI.
Tratamiento de insuficiencia suprarenal aguda
Los pacientes con insuficiencia suprarrenal aguda requieren rehidratación inmediata, por lo general con solución salina a velocidades iniciales de 1 L/h, y vigilancia cardiaca continua. La restitución con glucocorticoides debe iniciarse con una inyección en bolo de 100 mg de hidrocortisona, seguida de la administración de 200 mg de hidrocortisona en 24 h, en infusión continua o con inyecciones en bolo IV o IM. La restitución de mineralocorticoides tiene que iniciarse cuando la dosis diaria de hidrocortisona haya disminuido a menos de 50, mg porque las dosis altas de esta brindan suficiente estimulación de los receptores de mineralocorticoides.
La restitución de glucocorticoides para el tratamiento de la insuficiencia suprarrenal crónica debe administrarse en dosis que sustituyan la producción fisiológica diaria de cortisol, lo cual por lo general se logra con la administración oral de 15 a 25 mg de hidrocortisona en dos o tres dosis divididas. El embarazo exige un aumento de 50% en la dosis de hidrocortisona durante el último trimestre. En todos los sujetos tiene que administrarse al menos la mitad de la dosis diaria en la mañana, al despertar. No se prefieren los glucocorticoides de larga acción, como la prednisolona o la dexametasona, porque ocasionan una exposición aumentada a glucocorticoides por la activación prolongada de receptores de glucocorticoides en periodos de secreción fisiológicamente baja de cortisol. No hay equivalencias de dosis bien establecidas, pero, como guía, es posible asumir potencias equivalentes con 1 mg de hidrocortisona, 1.6 mg de acetato de cortisona, 0.2 mg de prednisolona, 0.25 mg de prednisona y 0.025 mg de dexametasona. Las preparaciones de glucocorticoides estándar disponibles en la actualidad no logran imitar el ritmo fisiológico de secreción de cortisol (fig. 3). Sin embargo, esto puede superarse con preparaciones de hidrocortisona de liberación modificada recientes, que han tenido resultados prometedores en el tratamiento de pacientes con insuficiencia suprarrenal primaria por hiperplasia suprarrenal congénita.
La vigilancia del remplazo de glucocorticoides se basa sobre todo en los antecedentes y la exploración de signos y síntomas sugestivos de administración excesiva o insuficiente del fármaco, lo que incluye la valoración del peso corporal y de la presión sanguínea. La ACTH plasmática, el cortisol libre en orina de 24 h o las curvas diarias de cortisol en suero muestran si se tomó o no la hidrocortisona, pero no ofrecen información confiable sobre la calidad de la restitución. En individuos con insuficiencia suprarrenal primaria aislada, la vigilancia debe incluir estudios de detección de enfermedad tiroidea autoinmunitaria, y las mujeres deben estar al tanto de la posibilidad de desarrollar insuficiencia ovárica prematura. El tratamiento suprafisiológico de glucocorticoides con dosis equivalentes a 30 mg de hidrocortisona o más, afecta el metabolismo óseo y estos sujetos deben someterse a una valoración regular de la densidad mineral ósea. Todas las personas con insuficiencia suprarrenal necesitan saber de la necesidad de ajustar la dosis de glucocorticoides en situaciones de estrés. En general, estas correcciones consisten en duplicar la dosis oral habitual de glucocorticoides en caso de enfermedades concomitantes con fiebre y reposo en cama, así como la necesidad de inyectar de inmediato 100 mg de hidrocortisona IV o IM, seguidos de una venoclisis de 200 mg de hidrocortisona/24 h en casos de vómito prolongado, cirugía o traumatismo. Todos los pacientes, pero en particular los que viven en regiones con acceso restringido a servicios de salud, o viajan a ellas, deben llevar un equipo de emergencia para autoinyectarse hidrocortisona, además de sus brazaletes y tarjetas de emergencia de uso de esteroides, y deben recibir capacitación sobre cómo utilizarlos.
La restitución de mineralocorticoides en individuos con insuficiencia suprarrenal primaria se inicia con una dosis de 100 a 150 mcg de fludrocortisona. Para evaluar la adecuación del tratamiento, se toma la presión arterial, en posición sedente y de pie, a fin de detectar una disminución postural que sugiera hipovolemia. Además, deben medirse con frecuencia el sodio y el potasio séricos y la renina plasmática. Las concentraciones de renina deben mantenerse en los límites de referencia altos normales. Los cambios de dosis de glucocorticoides también pueden afectar la restitución de mineralocorticoides, porque el cortisol también se une a los receptores de mineralocorticoides; 40 mg de hidrocortisona equivalen a 100 mcg de fludrocortisona. Es importante advertir que la prednisona y la prednisolona tienen actividad mineralocorticoide reducida y la dexametasona no posee ninguna. En pacientes que viven o viajan a regiones con condiciones climáticas calurosas o tropicales, la dosis de fludrocortisona debe aumentarse de 50 a 100 mcg durante el verano. También es necesario ajustar las dosis de mineralocorticoides durante el embarazo, por la actividad antimineralocorticoide de la progesterona, pero se necesita con menos frecuencia que la corrección de la dosis de hidrocortisona. La renina plasmática no sirve como herramienta de vigilancia durante el embarazo, porque la renina aumenta de manera fisiológica durante la gestación.
La restitución de andrógenos suprarrenales es una opción en pacientes con falta de energía, a pesar de la restitución óptima de glucocorticoides y mineralocorticoides. También puede indicarse en mujeres con características de deficiencia de andrógenos, lo que incluye pérdida de la libido. La restitución de andrógenos suprarrenales puede lograrse con 25 a 50 mg de DHEA una vez al día. El tratamiento se supervisa con la cuantificación de DHEAS, androstenediona, testosterona y globulina transportadora de hormonas sexuales (SHBG, sex hormone-binding globulin), 24 h después de la última dosis de DHEA.
Insuficiencia suprarrenal inducida por glucocorticoides
Un posible efecto indeseable del tratamiento con glucocorticoides es la supresión del eje hipotálamo-hipófisis-glándulas suprarrenales. La insuficiencia suprarrenal inducida por glucocorticoides (GI-AI, glucocorticoid-induced adrenal insufficiency) puede observarse con cualquier ruta de administración exógena: oral, tópica, inhalada, intranasal e intraarticular. Los factores que influyen sobre el riesgo de GI-AI incluyen la duración de la terapia con glucocorticoides, el modo de administración, la dosis y la potencia del glucocorticoide en cuestión, el uso de fármacos concomitantes que interfieren con el metabolismo de los glucocorticoides y la susceptibilidad individual. Hasta la mitad de los usuarios de glucocorticoides orales a largo plazo (0.5% a 1.8% de la población general en países occidentales) desarrolla GI-AI: los individuos tratados con dosis suprafisiológicas durante más de tres o cuatro semanas tienen el riesgo mayor, mientras que la terapia a corto plazo por lo general es segura. La GI-AI ocurre en alrededor de 8% de los usuarios de glucocorticoides inhalados, pero el riesgo incrementa de manera sustancial (21–27%) cuándo se utilizan dosis mayores y durante más de un año. La GI-AI también se ha documentado en casi 50% de los pacientes que reciben inyecciones intraarticulares de esteroides, pero las evidencias en esta población son muy limitadas. El uso de fórmulas que contienen múltiples glucocorticoides y la administración concomitante de inhibidores potentes del citocromo P450 3A4 (que incluyen muchos antibióticos, antifúngicos y ritonavir, un inhibidor de proteasas), son factores que incrementan de manera dramática el riesgo de desarrollar GI-AI. El ritonavir es el fármaco ofensor que se reporta con mayor frecuencia, y se usa en combinaciones antivirales para tratar infecciones por los virus de la inmunodeficiencia humana (VIH), la hepatitis C y la COVID-19. Otros fármacos que incrementan el riesgo son los opioides, que también pueden alterar la respuesta del eje HPA.
Los médicos que prescriben glucocorticoide deben educar a sus pacientes sobre los diferentes aspectos del tratamiento con glucocorticoides, incluyendo el riesgo de GI-AI. Si los glucocorticoides ya no son necesarios para controlar la enfermedad subyacente y el tratamiento dura menos de tres a cuatro semanas, es posible interrumpir de manera abrupta la administración de glucocorticoides. En cambio, las personas bajo tratamiento a largo plazo deben reducir las dosis de manera gradual hasta acercarse a una dosis fisiológica (p. ej., 3–5 mg de prednisona). Los pacientes que desarrollan dependencia de dosis suprafisiológicas de glucocorticoides pueden presentar síntomas de abstinencia durante la reducción, incluyendo malestar general, fatiga, náusea, mialgias, artralgias y trastornos del sueño y el estado de ánimo. los síntomas pueden ser graves y los individuos en ocasiones se benefician de un incremento temporal de las dosis de glucocorticoides y una reducción subsiguiente más lenta de las dosis. Después de lograr una dosis de glucocorticoides fisiológica, es necesario evaluar la recuperación del eje HPA a través de una medición de cortisol sérico al iniciar la mañana, lo cual proporciona información sobre la probabilidad de desarrollar GI-AI. No hay límites de cortisol establecidos, los cuales son influenciados por el ensayo utilizado y otros factores, que incluyen estrés concurrente representativo y niveles de CBG alterados que generan niveles de cortisol falsamente elevados (p. ej., estrógenos orales y embarazo) o falsamente reducidos (p. ej., cirrosis hepática avanzada y síndrome nefrótico). Los lineamientos internacionales actuales indican que un nivel de cortisol al iniciar la mañana < 150 nmol/L (o 5 mcg/dl) sugiere de manera contundente GI-AI, mientras que valores > 300 nmol/L (o 10 mcg/dl) concuerdan con recuperación del eje HPA. Para valores indeterminados puede considerarse la estimulación con cosintropina. Muchos doctores no miden de manera rutinaria los niveles de cortisol en sujetos que reducen sus dosis de glucocorticoides; en dichos casos, las personas deben educarse y monitorizarse de manera estrecha para identificar manifestaciones clínicas de insuficiencia suprarrenal y debe agendarse de manera oportuna una evaluación bioquímica si estas se presentan.
Los pacientes con GI-AI establecida deben tratarse como cualquier otro paciente con insuficiencia suprarrenal secundaria. Además de la restitución diaria de glucocorticoides, los individuos deben recibir dosis de cobertura de estrés cuando se exponen a situaciones estresantes para evitar insuficiencia suprarrenal aguda. La terapia con mineralocorticoides no es necesaria por la producción preservada de aldosterona.
Hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita (CAH, congenital adrenal hyperplasia) la ocasionan mutaciones en genes que codifican enzimas esteroidogénicas que participan en la síntesis de glucocorticoides (CYP21A2, CYP17A1, HSD3B2 y CYP11B1) o en el cofactor enzimático P450 oxidorreductasa que sirve como un donador de electrones para CYP21A2 y CYP17A1 (fig. 1). De manera invariable, los sujetos afectados por CAH presentan deficiencia de glucocorticoides. Dependiendo del paso exacto en el que ocurre el bloqueo enzimático, también puede ocurrir producción excesiva de mineralocorticoides o deficiencia en la producción de esteroides sexuales (cuadro 10). El diagnóstico de CAH se establece con facilidad midiendo los esteroides acumulados antes del bloqueo enzimático, en el suero o la orina, de preferencia mediante ensayos basados en espectrometría de masas (cuadro 10).
|
VARIANTE |
GEN |
IMPACTO EN LA SÍNTESIS DE ESTEROIDES |
ESTEROIDES MARCADORES DE DIAGNÓSTICO EN EL SUERO (Y LA ORINA) |
|
Deficiencia de 21-hidroxilasa (21OHD) |
CYP21A2 |
Deficiencia de glucocorticoides, deficiencia de mineralocorticoides y exceso de andrógenos suprarrenales |
17-hidroxiprogesterona, 21-desoxicortisol (pregnanetriol, 17-hidroxipregnenolona y pregnanetriolona) |
|
Deficiencia de 11β-hidroxilasa (11OHD) |
CYP11B1 |
Deficiencia de glucocorticoides, exceso de mineralocorticoides y exceso de andrógenos suprarrenales |
11-desoxicortisol, 11-desoxicorticosterona (tetrahidro-11-desoxicortisol y tetrahidro-11-desoxicorticosterona) |
|
Deficiencia de 17α-hidroxilasa (17OHD) |
CYP17A1 |
(Deficiencia de glucocorticoides) exceso de mineralocorticoides y deficiencia de andrógenos |
11-desoxicorticosterona, corticosterona, pregnenolona, progesterona (tetrahidro-11-desoxicorticosterona, tetrahidrocorticosterona, pregnenediol, pregnanediol) |
|
Deficiencia de deshidrogenasa DE 3β-hidroxiesteroide (3bHSDD) |
HSD3B2 |
Deficiencia de glucocorticoides (deficiencia de mineralocorticoides), exceso de andrógenos suprarrenales (mujeres y varones) y deficiencia de andrógenos gonadales (varones) |
17-hidroxipregnenolona (pregnanetriol) |
|
Deficiencia de P450 oxidorreductasa (PORD) |
POR |
Deficiencia de glucocorticoides (exceso de mineralocorticoides), exceso de andrógenos prenatales, deficiencia de andrógenos posnatales y malformaciones esqueléticas |
Pregnenolona, progesterona, 17-hidroxiprogesterona (pregnanediol, pregnanetriol) |
Cuadro 10. Variantes de hiperplasia suprarrenal congénita.
Las mutaciones en CYP21A2 son la causa más prevalente de CAH y causan 90% a 95% de los casos. La deficiencia de 21-hidroxilasa altera la síntesis de glucocorticoides y mineralocorticoides (fig. 1), con disminución de la retroalimentación negativa a través del eje HPA. Esto lleva a una mayor liberación hipofisaria de ACTH, que dirige el aumento de la síntesis de precursores de andrógenos suprarrenales y el consiguiente exceso de andrógenos. El grado de alteración en la secreción de glucocorticoides y mineralocorticoides depende de la gravedad de las mutaciones. Las principales mutaciones de pérdida de función resultan en deficiencia combinada de glucocorticoides y mineralocorticoides (CAH clásica, de presentación neonatal), en tanto que las mutaciones menos graves solo afectan la síntesis de glucocorticoides (CAH virilizante simple, de presentación neonatal o en la infancia temprana). Las mutaciones más leves ocasionan el fenotipo clínico menos grave, la hiperplasia suprarrenal congénita no clásica, que suele manifestarse en la adolescencia y el comienzo de la vida adulta, con producción de glucocorticoides conservada.
El exceso de andrógenos está presente en todos los pacientes y se manifiesta con una amplia variedad fenotípica, que va desde virilización grave de los genitales externos en niñas recién nacidas (p. ej., trastorno de desarrollo sexual 46,XX [DSD, disordered sex development]) hasta hirsutismo y oligomenorrea que simulan el fenotipo de síndrome de ovarios poliquísticos en mujeres jóvenes con CAH no clásica. En países que no realizan detección neonatal de CAH, los varones con CAH clásica por lo general se presentan con crisis suprarrenales que ponen en riesgo la vida, en las primeras semanas de vida (crisis de pérdida de sal); un genotipo virilizante simple se manifiesta en la infancia temprana con seudopubertad precoz y edad ósea avanzada, mientras que los varones con CAH no clásica por lo general se detectan solo mediante cribado familiar.
El tratamiento con glucocorticoides es más complejo que el de otras causas de insuficiencia suprarrenal primaria, porque no solo se necesita restituir los glucocorticoides faltantes, sino también controlar la ACTH aumentada y el consiguiente exceso de andrógenos. El tratamiento actual se complica por la falta de preparaciones de glucocorticoides que imiten el perfil de secreción diurna de cortisol, lo que resulta en un periodo prolongado de estimulación con ACTH y la producción subsecuente de andrógenos durante las primeras horas de la mañana. En la infancia, optimizar el desarrollo puberal y el crecimiento son metas importantes del tratamiento con glucocorticoides, además de prevenir las crisis suprarrenales y el tratamiento de DSD 46,XX. En adultos, el objetivo cambia a preservar la fertilidad y prevenir los efectos secundarios del exceso de tratamiento con glucocorticoides, a saber, el síndrome metabólico y la osteoporosis. La fertilidad puede comprometerse en mujeres por la presencia de oligomenorrea/amenorrea con anovulación crónica como consecuencia del exceso de andrógenos. Los varones pueden desarrollar tejido testicular suprarrenal residual (TART, testicular adrenal rest tissue) (fig. 17) formado por células hiperplásicas con características suprarrenales y gonadales compartidas, localizado en la red testicular, y no debe confundirse con tumores testiculares. El TART es capaz de alterar la producción de espermatozoides e inducir fibrosis testicular que puede ser irreversible.
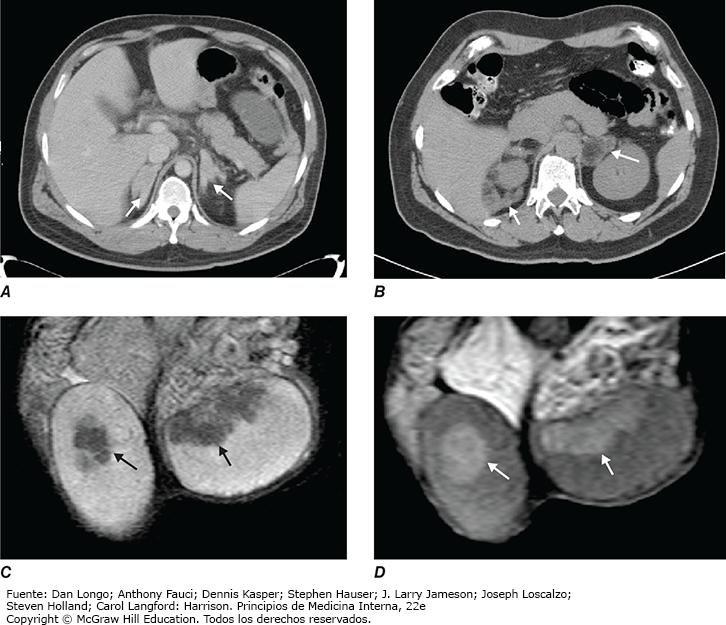
Tratamiento de la hiperplasia suprarrenal congénita
La hidrocortisona es una buena opción de tratamiento para evitar las crisis suprarrenales, pero puede necesitarse prednisolona de acción más prolongada para controlar el exceso de andrógenos. En niños, la hidrocortisona se administra en dosis divididas de 1 a 1.5 veces la tasa de producción normal de cortisol (~10 a 13 mg/m2 por día). En adultos, si no es suficiente la hidrocortisona, pueden administrarse glucocorticoides de acción intermedia (p. ej., prednisona), utilizando la menor dosis necesaria para suprimir el exceso de producción de andrógenos. Para lograr la fertilidad, a veces se necesita tratamiento con dexametasona, pero solo deberá administrarse durante el periodo más corto posible, para limitar los efectos adversos sobre el metabolismo. La introducción reciente de hidrocortisona de liberación prolongada y modificada, que simula el patrón endógeno fisiológico de liberación de cortisol, es prometedora, proporcionando un control efectivo del exceso de precursores de esteroides mientras que la dosis diaria de hidrocortisona es menor que la necesaria de hidrocortisona de liberación inmediata. El crinecerfont, un antagonista oral del factor liberador de corticotropina, ha demostrado eficacia para reducir las dosis de restitución de glucocorticoides necesarias para suprimir los andrógenos suprarrenales
La monitorización bioquímica debe incluir androstenediona y testosterona, y debe tener el objetivo de alcanzar el rango de referencia normal específico para cada sexo. La 17-hidroxiprogesterona (17OHP, 17-hydroxyprogesterone) es un marcador útil para determinar un tratamiento excesivo, señalado por concentraciones de 17OHP dentro del rango normal de controles sanos. El tratamiento excesivo con glucocorticoides puede suprimir el eje hipotálamo-hipófisis-gónada. Por lo tanto, el tratamiento necesita ajustarse con cuidado para evitar características clínicas del control de la enfermedad. Las dosis de estrés de glucocorticoides deben ser del doble o el triple de la dosis diaria, en casos de cirugías, enfermedades agudas o traumatismos graves. La CAH mal controlada puede ocasionar hiperplasia corticosuprarrenal, que le da el nombre a la enfermedad, y puede presentarse como hiperplasia macronodular posterior al exceso prolongado de ACTH (fig. 17). Las áreas nodulares pueden desarrollar producción autónoma de andrógenos suprarrenales y no responder al tratamiento con glucocorticoides. La prevalencia de suprarrenomielolipomas incrementa en la CAH; estos son benignos pero pueden requerir intervención quirúrgica por falta de crecimiento autolimitado.
Las necesidades de mineralocorticoides cambian durante la vida y son mayores en los niños, lo que se explica por la resistencia relativa a los mineralocorticoides que disminuye con la maduración continua de los riñones. Los niños con CAH por lo general reciben restitución de mineralocorticoides y sal. Sin embargo, los adultos jóvenes con CAH deben someterse a reevaluaciones de sus reservas de mineralocorticoides. La renina plasmática tiene que monitorizarse de manera regular y mantenerse en la mitad superior del rango normal de referencia.