El metabolismo cerebral implica tanto la producción como la utilización de energía; el catabolismo es la degradación y el anabolismo la síntesis de componentes y moléculas en las células. Para la formación de energía, el principal proceso catabólico es la degradación de la glucosa, que culmina en la formación de fosfato de alta energía en forma de adenosín trifosfato (ATP). Otros procesos catabólicos degradan proteínas estructurales y enzimáticas, lípidos y carbohidratos; estos procesos son necesarios para reemplazar las moléculas dañadas y disfuncionales. Estas moléculas se resintetizan mediante procesos anabólicos que renuevan las células y mantienen una función óptima. La función celular también requiere el mantenimiento de la homeostasis iónica, lo cual, en el caso de las neuronas, requiere una gran cantidad de energía. Los mecanismos fisiopatológicos de la lesión cerebral no se comprenden completamente, pero en última instancia representan un fallo de los procesos anabólicos para mantener la función celular normal. En este capítulo exploramos los posibles mecanismos de la lesión cerebral. Las causas del daño neuronal son multifacéticas, y una sola vía no puede explicar cómo se produce la lesión. Algunos mecanismos fisiopatológicos son comunes al daño causado por lesiones isquémicas, epileptogénicas y traumáticas, mientras que otros son específicos de cada uno de estos procesos. Esta revisión se centra en algunos desencadenantes comunes del daño neuronal, como las alteraciones en los gradientes iónicos, y explora cómo estos, a su vez, conducen a daños a largo plazo. También se analizan los fármacos y procedimientos clínicos que pueden contribuir a reducir el daño cerebral a largo plazo.
Metabolismo Cerebral
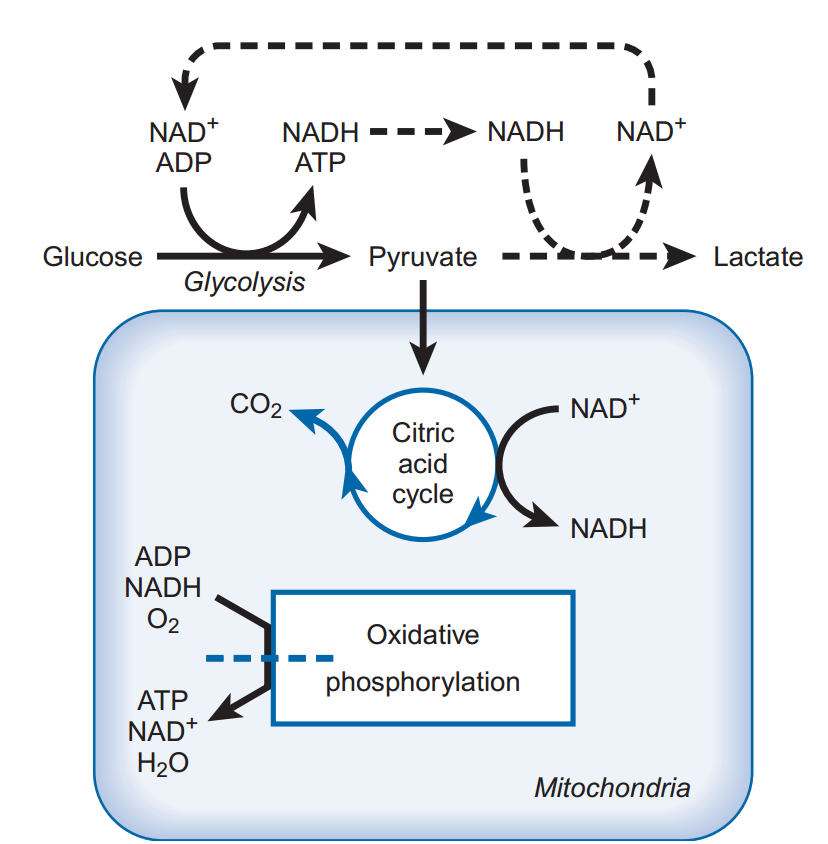
La principal sustancia utilizada para la producción de energía en el cerebro es la glucosa. Dado que la glucosa no atraviesa libremente la barrera hematoencefálica, requiere un transportador para entrar en el cerebro. Este transportador no requiere energía y solo puede mover la glucosa a favor de su gradiente de concentración, de una concentración mayor a una menor. Normalmente, los niveles de glucosa en sangre están bien regulados, por lo que las concentraciones de glucosa en el cerebro son adecuadas; sin embargo, si los niveles de glucosa en sangre disminuyen, el suministro de glucosa no puede satisfacer las necesidades energéticas del cerebro. Por lo tanto, unos niveles adecuados de glucosa en sangre son fundamentales para la actividad cerebral normal. Durante un choque insulínico u otras afecciones que provocan una reducción de la glucosa en sangre, la pérdida de consciencia puede ser consecuencia de una energía insuficiente debido a los bajos niveles de glucosa en el cerebro. Cuando los niveles de glucosa y oxígeno son suficientes, la glucosa se metaboliza a piruvato en la vía glucolítica (Fig. 1). Este proceso bioquímico genera ATP a partir de adenosín difosfato (ADP) y fosfato inorgánico, y produce nicotinamida adenina dinucleótido reducido (NADH) a partir de nicotinamida adenina dinucleótido (NAD+). El piruvato resultante de esta reacción ingresa al ciclo del ácido cítrico, que, en lo que respecta a la producción de energía, genera principalmente NADH a partir de NAD+. Las mitocondrias utilizan oxígeno para acoplar la conversión de NADH a NAD+ con la producción de ATP a partir de ADP y fosfato inorgánico. Este proceso, denominado fosforilación oxidativa, forma tres moléculas de ATP por cada NADH convertido y produce un máximo de 38 moléculas de ATP por cada molécula de glucosa metabolizada. Dado que numerosas partes de esta vía metabólica satisfacen otras necesidades metabólicas, como la síntesis de aminoácidos y la formación de equivalentes reductores para otras vías sintéticas, el rendimiento normal de esta vía energética es de aproximadamente 30 a 35 moléculas de ATP por cada molécula de glucosa. Esta vía requiere oxígeno; si no está presente, las mitocondrias no pueden producir ATP ni regenerar NAD+ a partir de NADH. El metabolismo de la glucosa requiere NAD+ como cofactor y se bloquea en su ausencia. Por lo tanto, en ausencia de oxígeno, la glucólisis se lleva a cabo mediante una vía modificada denominada glucólisis anaeróbica. Esta modificación implica la conversión de piruvato a lactato, regenerando NAD+. Este proceso produce iones de hidrógeno, lo que puede acentuar el daño neuronal si el pH intracelular disminuye. Un problema importante de la glucólisis anaeróbica, además de la disminución del pH, es que solo se forman dos moléculas de ATP por cada molécula de glucosa metabolizada. Este nivel de producción de ATP es insuficiente para satisfacer las necesidades energéticas del cerebro. Además, la isquemia reduce el suministro de glucosa, por lo que incluso la glucólisis anaeróbica se bloquea. Cuando se reduce el suministro de oxígeno a una neurona, los mecanismos que reducen o ralentizan la disminución de los niveles de ATP incluyen: (1) la utilización de las reservas de fosfocreatina (un fosfato de alta energía que puede donar su energía para mantener los niveles de ATP), (2) la producción de ATP a bajos niveles mediante la glucólisis anaeróbica y (3) una rápida cesación de la actividad electrofisiológica espontánea.
Procesos Celulares Que Requieren Energía
El bombeo de iones a través de la membrana celular es el principal requerimiento energético del cerebro. Las concentraciones de sodio, potasio y calcio en una neurona se mantienen frente a grandes gradientes electroquímicos con respecto al exterior celular. Cuando se mencionan el sodio (Na), el calcio (Ca) y el potasio (K) a lo largo del capítulo, nos referimos a su forma iónica (Na+, Ca++ y K+); esta es la única forma de estos compuestos presente en las células vivas. Cuando una neurona no está excitada, se producen fugas lentas de potasio hacia fuera de la célula y de sodio hacia dentro. El potencial de reposo de una neurona depende principalmente del potencial de equilibrio electroquímico del potasio, que en la mayoría de las neuronas es de aproximadamente -94 mV. Existe cierta permeabilidad al sodio y al calcio, por lo que el potencial de reposo de una neurona suele estar entre -60 y -70 mV. Debido a que el potencial de membrana celular no es igual al potencial de equilibrio de un ion, se produce una fuga de iones a través de sus gradientes electroquímicos. Si esta fuga no se corrigiera mediante bombas iónicas dependientes de energía, el potencial de membrana caería a 0 mV y la célula se despolarizaría y moriría. Las bombas iónicas se dividen en dos categorías principales: (1) las que utilizan ATP directamente para bombear iones y (2) las que utilizan la energía del gradiente de Na para cotransportar otro ion o molécula. La energía final para estas últimas bombas proviene del ATP a través de la Na/K ATPasa, que transporta iones Na y mantiene el gradiente de energía de Na; ejemplos de estas bombas de intercambio incluyen los transportadores Na/Ca, Na/H y Na/glutamato. Ejemplos de la primera categoría de bombas son la Na/K ATPasa, principal consumidora de energía en las neuronas, y la Ca ATPasa. Las bombas iónicas primarias que utilizan directamente ATP son importantes porque establecen los gradientes electroquímicos necesarios para que las bombas secundarias, las bombas de intercambio iónico, funcionen en la dirección deseada. De hecho, durante la isquemia, estas bombas no disponen de la energía suficiente para operar, y esta condición es una causa principal de la despolarización neuronal y la muerte celular. La actividad neuronal aumenta notablemente el flujo de sodio, potasio y calcio al abrir los canales iónicos de Na, K y Ca; esta apertura incrementa la velocidad de bombeo iónico necesaria para mantener las concentraciones iónicas celulares normales. Dado que el bombeo iónico utiliza ATP como fuente de energía, el requerimiento de ATP de las neuronas activas es mayor que el de las neuronas no excitadas. Aproximadamente el 60 % de la energía que utiliza el cerebro se requiere para la actividad funcional, y el resto se utiliza para mantener la integridad celular. Los anestésicos reducen la actividad neuronal y, por lo tanto, el consumo de ATP por la actividad funcional, pero no reducen la energía necesaria para la integridad del cerebro. Si la producción de energía no satisface la demanda energética del cerebro, las neuronas primero se vuelven inexcitables y luego sufren daños irreversibles.
Las neuronas requieren energía para mantener su estructura y función interna. Las membranas, los orgánulos internos y el citoplasma de cada célula están compuestos de carbohidratos, lípidos y proteínas que requieren energía para su síntesis. Los canales iónicos, las enzimas y los componentes estructurales celulares son importantes moléculas proteicas que se forman, modifican y degradan continuamente en la célula. Si no hay ATP disponible, la síntesis de proteínas no puede continuar y la neurona muere. Los carbohidratos y los lípidos también se sintetizan y degradan continuamente en las neuronas que funcionan con normalidad; su metabolismo también requiere energía. La mayor parte de la síntesis celular tiene lugar en el cuerpo celular, y se requiere energía para el transporte de componentes a lo largo del axón hasta las terminaciones nerviosas. Por lo tanto, se requiere energía para mantener la integridad de las neuronas incluso en ausencia de actividad electrofisiológica.
Neuroantomía
El cerebro se diferencia regionalmente tanto estructural como funcionalmente; esta sección ofrece una visión general de la funcionalidad de las distintas regiones cerebrales. Esto es importante en relación con el ictus, ya que cuando una arteria se obstruye, la función de las neuronas en la región irrigada por dicha arteria se ve comprometida. Los detalles de la neuroanatomía y la neurofisiología del cerebro requerirían un libro aparte; dos obras recomendadas para un análisis detallado son Neuroanatomía Clínica, de R. Snell, y Neurofisiología y Principios de Neurociencia, de Kandel et al.
La corteza cerebral tiene cuatro lóbulos principales a cada lado: el frontal, el parietal, el occipital y el temporal (Fig. 2).Las vías sensoriales de un lado del cuerpo cruzan la línea media y proporcionan información a la corteza somatosensorial del lado opuesto. Las vías motoras que se originan en la corteza motora de un lado se decusan en el bulbo raquídeo, descienden por la médula espinal a través de los tractos corticoespinales laterales, hacen sinapsis con las motoneuronas ventrales en la sustancia gris de la médula y transmiten la respuesta motora al lado opuesto del cuerpo. La parte anterior del lóbulo frontal (área prefrontal) influye en la personalidad, la orientación, la concentración y el juicio; es importante para dirigir la actividad intelectual hacia un objetivo. La circunvolución precentral del lóbulo frontal es la corteza motora primaria, tiene proyección a las motoneuronas de la médula espinal y controla los movimientos finos. Las áreas de asociación premotora se localizan rostralmente y reciben información de otras áreas motoras del cerebro, como los ganglios basales, el cerebelo y el núcleo rojo. Por lo tanto, la corteza premotora y la motora son responsables de integrar la información de las áreas motoras de todo el cerebro, lo que da lugar a movimientos intencionados. Adyacente a la circunvolución precentral, al otro lado del surco central, se encuentra la circunvolución postcentral del lóbulo parietal; esta es la corteza somatosensorial primaria y recibe información sobre el tacto fino. Posterior a la circunvolución postcentral se encuentran las áreas de asociación somatosensorial, que ayudan a interpretar y analizar las sensaciones táctiles. Todas las áreas sensoriales primarias del cerebro tienen áreas de asociación sensorial que analizan e interpretan aún más estas señales. El lóbulo temporal se localiza debajo de los lóbulos frontal y parietal y contiene las áreas auditivas primarias y de asociación auditiva. Un hemisferio cerebral se considera dominante y un área en particular de este es importante para la interpretación del lenguaje y la producción del habla. Esta área se denomina área de Wernicke. El área de Wernicke es de vital importancia y las lesiones en ella provocan afasia profunda; generalmente se considera que incluye la parte posterior del giro temporal superior y el giro angular en el hemisferio cerebral dominante. El giro angular es un área de asociación multimodal importante en el lóbulo parietal, adyacente al lóbulo temporal. Las áreas de asociación multimodal analizan la información sensorial proveniente de áreas de asociación sensoriales individuales y proporcionan un análisis complejo de dicha información, determinando la respuesta a estímulos complejos. El área de Wernicke se delimita con sumo cuidado durante la neurocirugía y se evita dañarla siempre que sea posible. Está irrigada por la arteria cerebral media y se producen déficits profundos tras la oclusión de esta arteria debido a un accidente cerebrovascular isquémico. Las lesiones en esta área aíslan a la persona, impidiéndole comunicarse o comprender la comunicación verbal o escrita. Esta área activa directamente el área de Broca en el lóbulo frontal, un área premotora del lenguaje. Las lesiones en el lóbulo parietal del hemisferio no dominante provocan déficits visuoespaciales y heminegligencia (ignorar la mitad del espacio exterior).
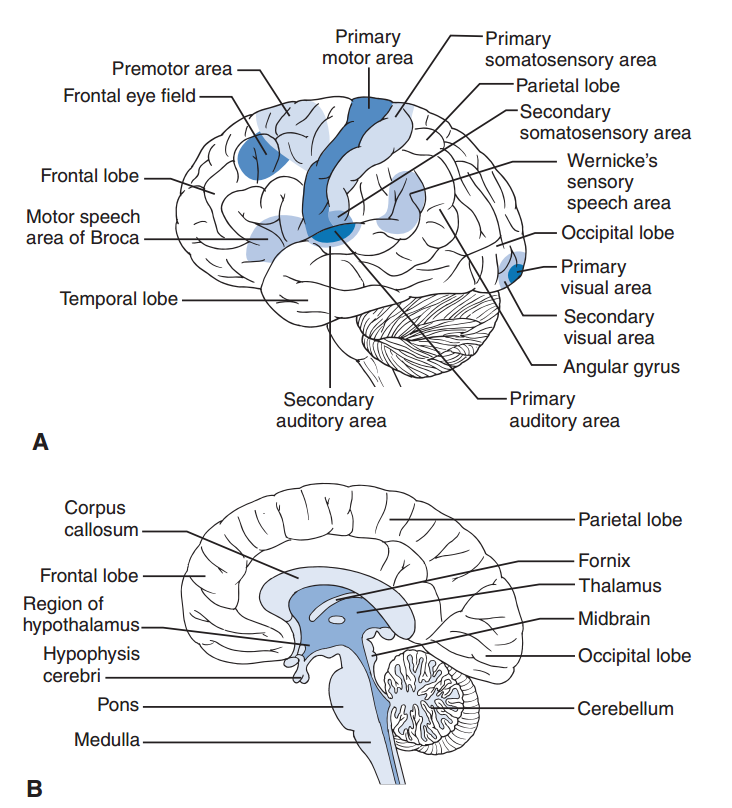
El tálamo se localiza medialmente y es un importante centro de relevo para la información que entra y sale de la corteza cerebral. El hipotálamo, situado debajo del tálamo, es importante para diversas funciones reguladoras del cuerpo, como el hambre, la sed y la regulación de la temperatura, e integra la actividad conductual y motivacional del sistema límbico con las respuestas autonómicas. El sistema límbico incluye la corteza límbica, el hipocampo y la amígdala, y está asociado con las sensaciones de recompensa y castigo, el comportamiento emocional, el aprendizaje y la memoria. El hipocampo y el lóbulo temporal medial son importantes para la formación de la memoria a largo plazo; la amígdala transmite el contenido emocional de la memoria. Los ganglios basales también se localizan medialmente a la corteza y son importantes para la función motora y el inicio de los movimientos. La enfermedad de Parkinson se debe a lesiones en la sustancia negra, un área dopaminérgica, que provocan temblor en reposo y bradicinesia. La demencia es una manifestación no motora de las enfermedades de los ganglios basales e indica que estas áreas, consideradas principalmente motoras, también pueden influir profundamente en el comportamiento.
El cerebelo se localiza sobre el tronco encefálico y desempeña un papel importante en la actividad motora rápida aprendida, así como en el control postural. Recibe información de la corteza motora y retroalimentación propioceptiva del cuerpo para comparar el movimiento deseado con el movimiento real producido por los músculos. Es importante saber que el cerebelo lateral no se cruza y controla el mismo lado del cuerpo; por ejemplo, el cerebelo derecho controla los músculos del lado derecho del cuerpo, que también está controlado por la corteza motora izquierda. De esta manera, la información de la corteza cerebelosa cruza la línea media en su camino hacia la corteza cerebral.
El tronco encefálico está formado por el mesencéfalo, la protuberancia anular y el bulbo raquídeo, y se continúa estructuralmente con la médula espinal. Los nervios craneales III a XII se originan en el tronco encefálico o en sus núcleos. Vías neuronales ascendentes y descendentes recorren esta parte del cerebro y hacen sinapsis con las neuronas que contiene; esta área alberga la formación reticular y el sistema reticular activador, responsables del estado de alerta y la consciencia. Esta área es fundamental para el control de la presión arterial, la frecuencia cardíaca, la respiración, la deglución y otras funciones corporales. Las lesiones en esta zona pueden provocar coma o muerte súbita.
La médula espinal permite la comunicación entre el cerebro y el cuerpo, y contiene vías sensoriales ascendentes y vías motoras descendentes. Los tractos espinotalámicos anterolaterales transmiten el tacto grueso, la temperatura y el dolor; penetran en la sustancia gris de la médula y hacen sinapsis en el asta dorsal. Los axones de las neuronas postsinápticas cruzan la línea media y ascienden por la médula espinal hasta el tronco encefálico y el tálamo a través de los tractos anterolaterales. Las columnas dorsales transmiten el tacto fino y la propiocepción, y ascienden por la médula del mismo lado del cuerpo, cruzando la línea media tras hacer sinapsis en los núcleos del bulbo raquídeo. El destino final de estos axones es el tálamo, desde donde la información se transmite a la corteza somatosensorial en la circunvolución poscentral. Los axones de las columnas dorsales también envían ramificaciones a la médula espinal, a la altura o cerca del nivel en el que entran. La médula espinal posee circuitos neuronales que modifican la información que llega al cerebro y también median reflejos locales como la respuesta al dolor y el control de la tensión y el tono muscular.
Fisiopatología
Isquemia
Cuando se limita el riego sanguíneo al cerebro, puede producirse daño isquémico en las neuronas; el cerebro es el órgano más sensible a este daño. La zona del cerebro correspondiente al territorio de la arteria cerebral bloqueada determina qué funciones se alteran o se pierden tras la isquemia focal; esto se corresponde con la anatomía funcional descrita en la sección anterior. Las neuronas en las zonas isquémicas se dañan por la pérdida de energía; el resto de esta sección describe los eventos celulares posteriores a la isquemia que conducen a este daño.
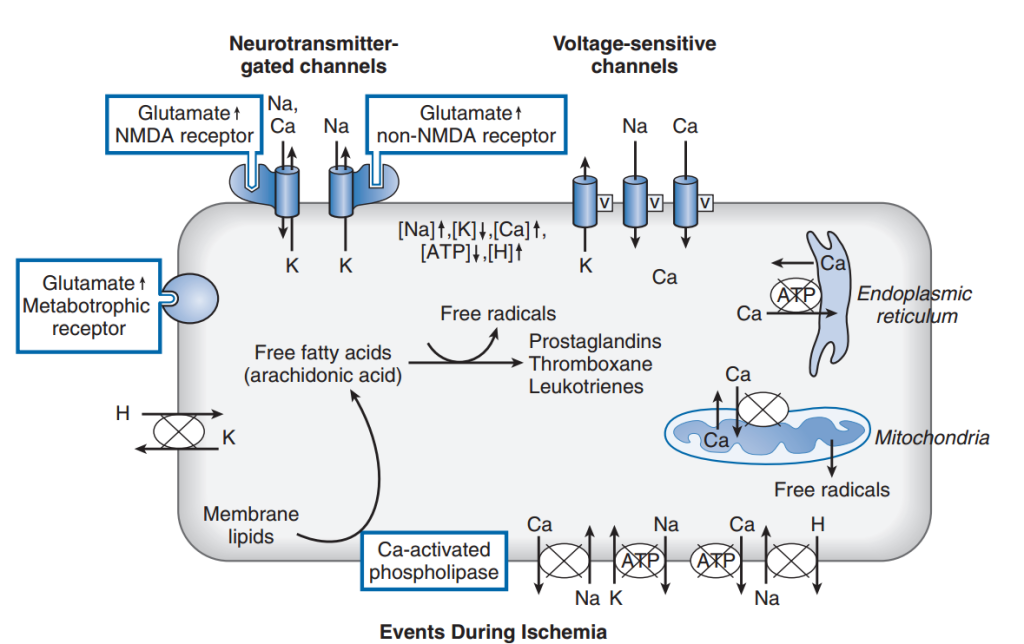
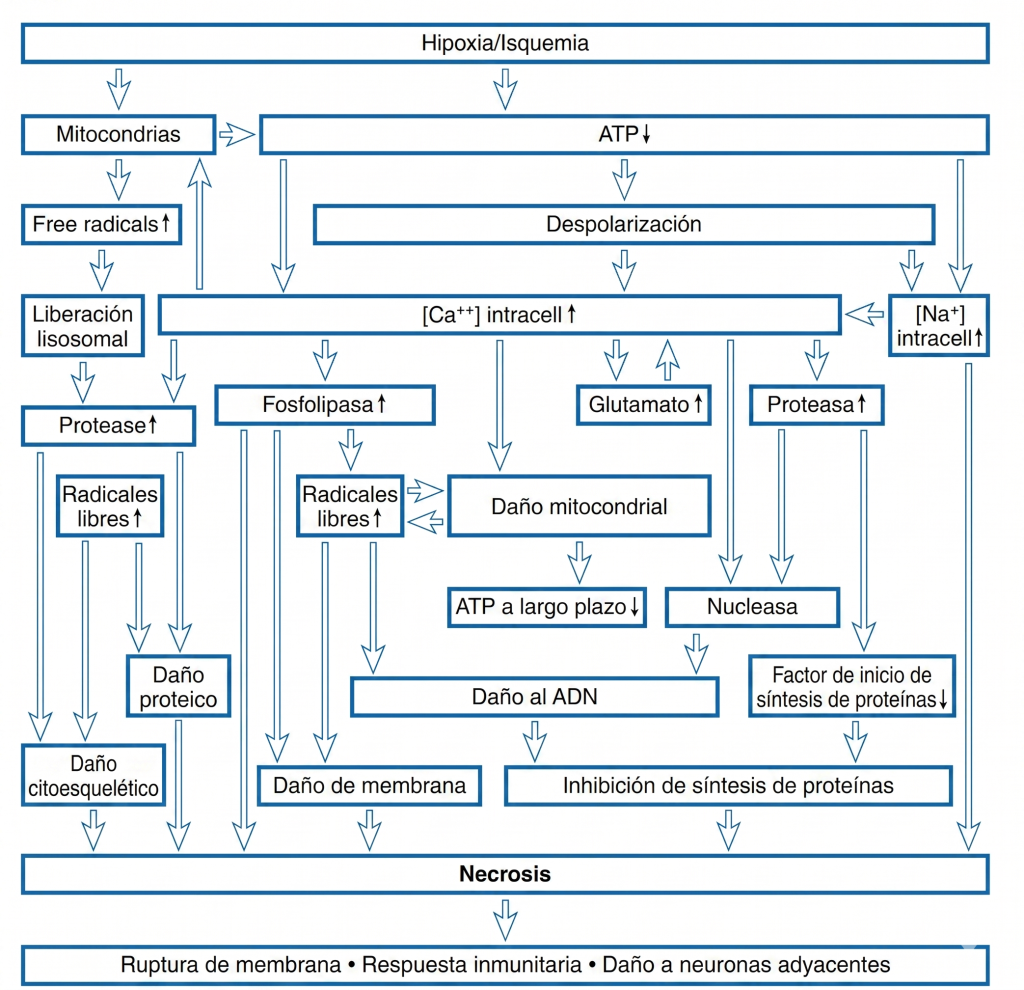
El evento central que desencadena el daño por hipoxia o isquemia es la reducción de la producción de energía debido al bloqueo de la fosforilación oxidativa. Esto provoca una disminución del 95 % en la producción de ATP por molécula de glucosa. A esta tasa de producción, los niveles de ATP disminuyen, lo que conlleva la pérdida de los mecanismos homeostáticos dependientes de la energía. Además, durante la isquemia se interrumpe el suministro de glucosa, así como la eliminación de metabolitos. La actividad de las bombas iónicas dependientes de ATP se reduce y los niveles intracelulares de sodio y calcio aumentan, mientras que los de potasio disminuyen (Fig. 3). Estos cambios iónicos provocan la despolarización de las neuronas y la liberación de aminoácidos excitatorios como el glutamato. Asimismo, el glutamato se libera de las neuronas debido a la inversión del transportador de glutamato, que bombea glutamato al compartimento extracelular cuando se alteran los gradientes de iones de sodio y potasio celulares. Los altos niveles de glutamato despolarizan aún más las neuronas al activar los receptores AMPA (α-amino-3-hidroxi-5-metil-4-isoxazolpropionato) y NMDA (N-metil-D-aspartato), lo que aumenta la conductancia de iones sodio y potasio. El receptor NMDA también permite la entrada de calcio, desencadenando vías dañinas adicionales. El glutamato activa los receptores metabotrópicos, que, a través de sistemas de segundos mensajeros, pueden aumentar la liberación de calcio de los depósitos intracelulares y activar otros procesos bioquímicos. El daño causado por el exceso de glutamato se denomina excitotoxicidad y se debe a la activación de los receptores de glutamato y a los cambios iónicos y bioquímicos que la acompañan.
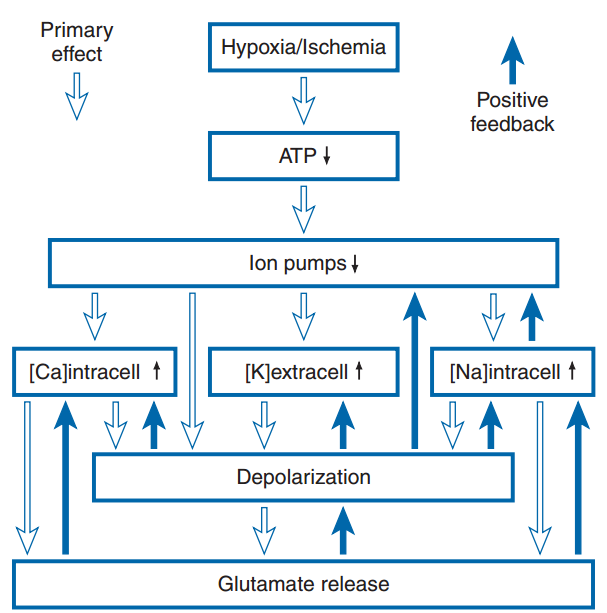
Además del aumento del influjo a través de los canales de membrana, el calcio citosólico se incrementa debido a la reducción del bombeo de calcio desde la célula y a la mayor liberación de calcio de orgánulos intracelulares como el retículo endoplasmático (Fig. 4). Se cree que el alto nivel de calcio citoplasmático desencadena una serie de eventos que conducen al daño isquémico. Estos incluyen el aumento de la actividad de proteasas y fosfolipasas. Las fosfolipasas elevan los niveles de ácidos grasos libres, como el ácido araquidónico, y de radicales libres. Los radicales libres también se generan por la oxidación mitocondrial incompleta. Uno de los radicales libres más dañinos es el peroxinitrito, que se forma por la combinación de óxido nítrico y otro radical libre. Se sabe que los radicales libres dañan las proteínas y los lípidos, mientras que los ácidos grasos libres interfieren con la función de la membrana.
Durante la isquemia se produce una acumulación de lactato e iones de hidrógeno, lo que disminuye el pH intracelular y puede dar lugar a la formación de radicales libres. Todos estos procesos, junto con la menor capacidad para sintetizar proteínas y lípidos, contribuyen al daño irreversible que se produce con la isquemia (Box 1).
Desencadenantes
- Trifosfato de adenosina (ATP) ↓
- Potasio extracelular ↑
- Sodio intracelular ↑
- Calcio intracelular ↑
- Niveles de radicales libres ↑
- Despolarización ↑
- Nivel de glutamato ↑
Efectores
- Actividad de proteasas ↑
- Acción de radicales libres ↑
- Daño del ADN ↑
- Actividad de fosfolipasas ↑
- Factores mitocondriales ↑ (citocromo c → activación de caspasas)
Cambios Funcionales Críticos
- Daño mitocondrial ↑
- Activación de la cascada apoptótica ↑
- Factores antiapoptóticos ↓
- Daño proteico ↑
- Síntesis de proteínas ↓
- Daño del citoesqueleto ↑
Etapa Final
- Apoptosis ↑ (muerte celular programada)
- Necrosis ↑ (desintegración celular)
Adaptado de: Lipton P. Muerte celular isquémica en neuronas cerebrales. Physiol Rev. 1999;79:1431–1568.
Además, la activación de la fosfolipasa produce un exceso de ácido araquidónico, que, tras la reoxigenación, puede formar eicosanoides, como tromboxano, prostaglandinas y leucotrienos. Estas sustancias pueden provocar una fuerte vasoconstricción, reducir el flujo sanguíneo en el periodo postisquémico, alterar la barrera hematoencefálica y aumentar la formación de radicales libres tras la reperfusión.
Los procedimientos que protegen contra el daño isquémico deben interferir con los cambios celulares producidos por la isquemia (Recuadro 2). Además de estos eventos desencadenantes directos, existe un daño a largo plazo que se hace evidente horas y días después del episodio isquémico. Parte de este daño tardío es necrótico y la lisis de las células provoca la activación de la microglía. Los linfocitos, las células polimorfonucleares y los macrófagos pueden invadir el sistema nervioso central, lo que conlleva un daño adicional. Aunque la activación del receptor de histamina se asocia generalmente con la activación del sistema inmunitario, el receptor de histamina implicado es el receptor H1. En el sistema nervioso central, el receptor H2 es el que se activa principalmente, y reduce los procesos inmunológicos y mejora la recuperación de la isquemia. El bloqueo de la activación del sistema inmunitario puede reducir el daño. Es evidente que también existe una muerte celular programada como resultado del episodio. Esta muerte celular programada apoptótica, similar a la que ocurre durante el desarrollo neuronal, puede continuar días después del episodio inicial.
Cambios Vasculares
- Vasoespasmo
- Estasis eritrocitaria (aglutinación de eritrocitos)
- Hipoperfusión
- Agregación plaquetaria
- Lesión endotelial
- Adhesión leucocito-endotelio
- Alteración de la barrera hematoencefálica
Cambios Neuronales
- Disminución del trifosfato de adenosina (ATP)
- Entrada de sodio
- Salida de potasio
- Acidosis intracelular
- Elevadas concentraciones intracelulares de calcio
- Proteasas activadas por calcio
- Activación de caspasas
- Activación de fosfolipasas
- Formación y degradación del ácido araquidónico
- Producción de radicales libres
- Liberación de aminoácidos excitatorios
- Alteración de los transportadores de iones y aminoácidos
- Autofagia
- Apoptosis
- Necrosis
Necrosis versus apoptosis
Existen dos procesos principales que conducen a la muerte neuronal. El primero, la necrosis, se debe a una lesión más grave que provoca la pérdida de la función mitocondrial; se caracteriza por la desintegración celular y la activación de la microglía y la respuesta inmunitaria. La respuesta inmunitaria y la inflamación activan y reclutan neutrófilos y macrófagos, que producen radicales libres y dañan las neuronas adyacentes. Este proceso aumenta el volumen y el tiempo de la lesión, lo que permite un daño neuronal continuo y extenso. En el segundo proceso, la apoptosis, la célula muere sin desintegrarse y no hay participación de la microglía ni del sistema inmunitario, con el potencial de causar un daño excesivo a las neuronas adyacentes. Este proceso suele retrasarse y puede conducir a la activación de genes de respuesta temprana inmediata (IEG), como c-Jun y c-Fos; se cree que estos genes afectan la expresión génica y conducen a la producción de proteínas apoptóticas o antiapoptóticas, que determinan si las neuronas sobrevivirán o morirán. Un conjunto de proteínas que conducen a la muerte neuronal son las cisteína proteasas, denominadas caspasas.
Estas enzimas se expresan como proenzimas, las cuales sufren un procesamiento proteolítico para generar enzimas activas que degradan proteínas importantes en la célula (Fig. 5). Existen vías intrínsecas y extrínsecas para activar las caspasas y la apoptosis. La Fig. 5 muestra la vía intrínseca activada por la liberación del citocromo c mitocondrial. Además, los receptores de muerte celular en la membrana neuronal pueden activarse por factores de muerte como el ligando Fas o el factor de necrosis tumoral, que activan directamente las caspasas. La vía apoptótica final hacia la muerte celular converge y es la misma para ambas vías, la intrínseca y la extrínseca. Se ha demostrado que el bloqueo de las caspasas inhibe la apoptosis. Dado que ahora se sabe que estas enzimas están presentes como proenzimas antes de la isquemia, no se requiere la síntesis de nuevas proteínas para inducir la apoptosis. Sin embargo, en ciertas condiciones se sintetizan proteínas proapoptóticas, y su síntesis puede provocar la muerte neuronal tardía. Se puede inducir otro conjunto de proteínas que bloquean la apoptosis y promueven la supervivencia neuronal después de la isquemia; ejemplos de estas proteínas son la proteína inhibidora de la apoptosis neuronal, las proteínas de choque térmico y ciertas proteínas antiapoptóticas de la familia Bcl-2. Por lo tanto, el destino de las neuronas isquémicas depende del equilibrio entre los procesos inhibidores y activadores de la apoptosis (Fig. 6). La síntesis de ciertos factores tróficos puede mejorar la supervivencia neuronal al inhibir la apoptosis (véase la Fig. 5). Se cree que la activación y liberación de ciertas citocinas, como el factor de necrosis tumoral y la interleucina-1β, son dañinas.
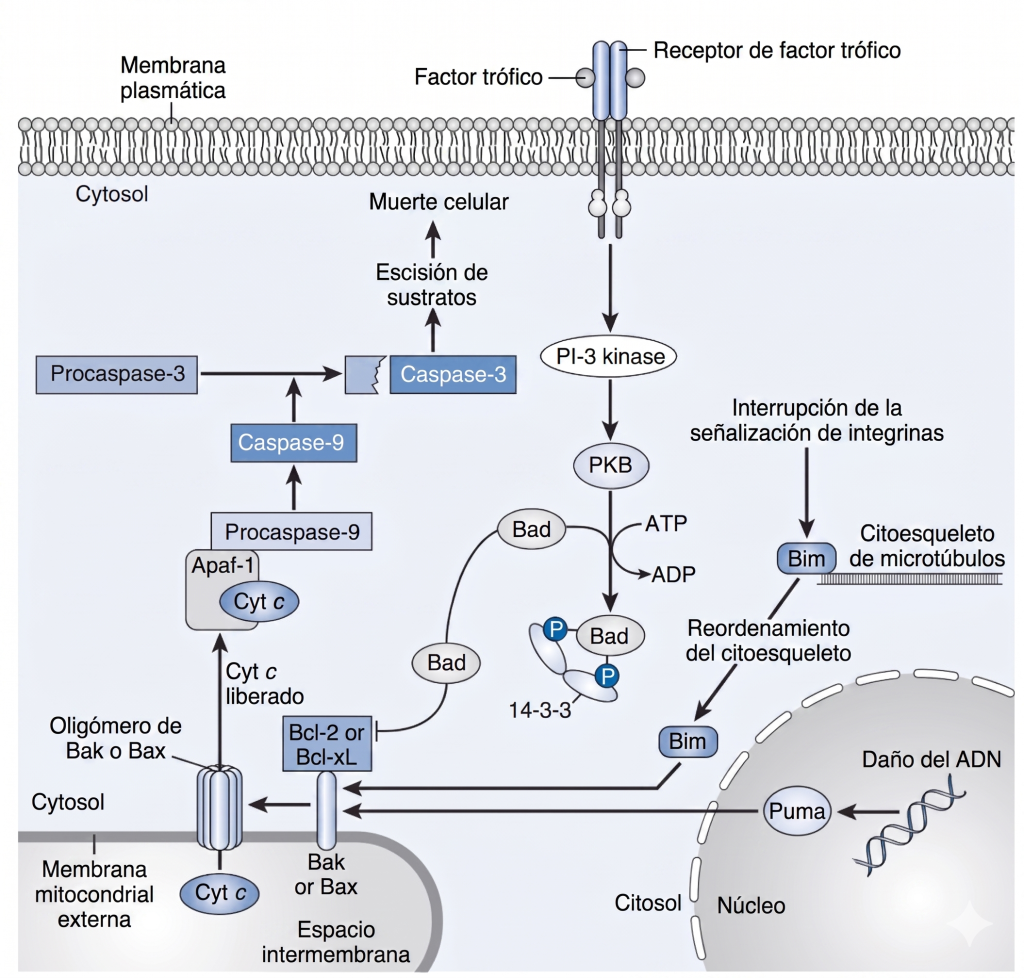
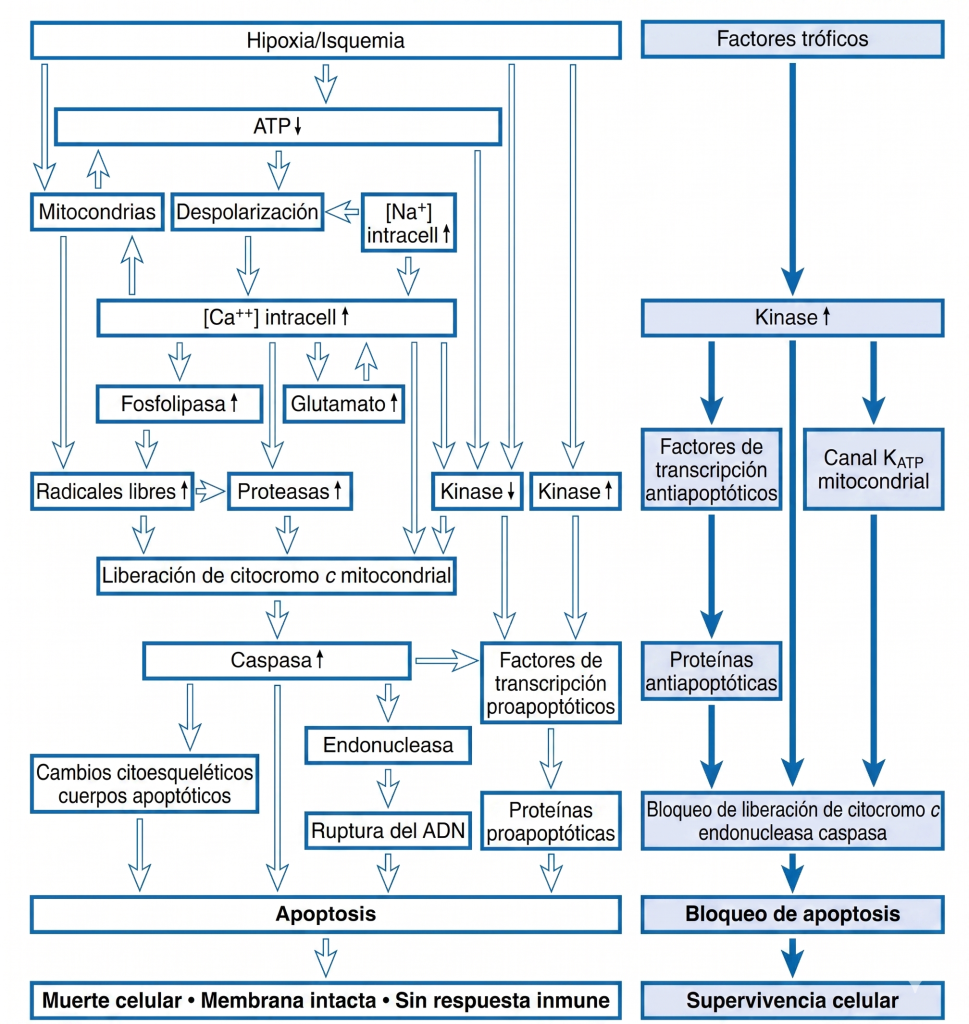
Así, se pueden contrastar la necrosis y la apoptosis, siendo la primera consecuencia de una isquemia más grave y provocando daños en el tejido adyacente (Fig. 7). La apoptosis es modulable, por lo que, una vez iniciada la vía apoptótica, las células tienen la posibilidad de ser rescatadas por sustancias tróficas (véase la Fig. 6).
Isquemia global versus isquemia focal
La isquemia puede ser global o focal; un ejemplo de la primera sería el paro cardíaco y, de la segunda, el accidente cerebrovascular localizado. Si bien los mecanismos que conducen al daño neuronal son probablemente similares para ambos tipos de isquemia, existen diferencias importantes entre ellos. En la isquemia focal, hay tres regiones. La primera, denominada núcleo isquémico, no recibe flujo sanguíneo y responde de la misma manera que el tejido isquémico global; la segunda, denominada penumbra, recibe flujo colateral y es parcialmente isquémica; la tercera región está perfundida normalmente. Si la lesión se mantiene durante un período prolongado, las neuronas de la penumbra mueren y el infarto (núcleo isquémico) aumenta de tamaño. Sobreviven más neuronas en la región de la penumbra si aumenta el flujo sanguíneo colateral o si se restablece la reperfusión de manera oportuna mediante la apertura del vaso bloqueado. En la isquemia global total, el tiempo hasta que se restablece la circulación es crítico, y solo los tiempos de isquemia muy cortos (del orden de minutos) son compatibles con la supervivencia. El daño neurológico selectivo tras la supervivencia a una isquemia global se debe principalmente a la sensibilidad diferencial de ciertas neuronas y regiones cerebrales. El hipocampo, especialmente la región de células piramidales CA1 (cornu ammonis 1), es extremadamente vulnerable al daño isquémico; la pérdida de aprendizaje y memoria es frecuente tras una isquemia global e hipoxia. Otras áreas con mayor sensibilidad a la isquemia global e hipoxia son el núcleo caudado y el putamen, así como ciertas áreas del cerebelo y la corteza cerebral.
Influencias genéticas en el daño neuronal
Los factores genéticos desempeñan un papel importante en la susceptibilidad de un individuo al accidente cerebrovascular isquémico. Tanto los factores ambientales (como la dieta y el estrés) como los genéticos se combinan para determinar el riesgo de accidente cerebrovascular. Un estudio realizado en la población islandesa reveló que los polimorfismos (cambios genéticos) en el locus genético ALOX5AP, que codifica la proteína activadora de la 5-lipoxigenasa, y PDE4D, que codifica la fosfodiesterasa 4D, aumentan la susceptibilidad al accidente cerebrovascular. Además, se ha observado que los polimorfismos de la apolipoproteína B y la apolipoproteína E incrementan la susceptibilidad al accidente cerebrovascular. Si bien los factores genéticos podrían influir en el riesgo neuronal, es más probable que aumenten el riesgo vascular, lo que se asocia con un incremento tanto en el accidente cerebrovascular como en la enfermedad cardíaca. Si se conociera la susceptibilidad genética de un paciente a las lesiones, sería posible elegir estrategias terapéuticas individualizadas para cada paciente con el fin de mejorar el resultado.
Se ha demostrado que los factores genéticos influyen en el riesgo cardiovascular, en particular con respecto a la hiperlipidemia; controlar estos factores con estatinas no solo reduce las enfermedades cardíacas, sino que también reduce las tasas de enfermedades cerebrovasculares y accidentes cerebrovasculares.
Posibles Tratamientos Para La Isquemia Cerebral
Estrategias de reperfusión
El objetivo del tratamiento del ictus isquémico es lograr la pronta restauración de la perfusión y preservar el tejido cerebral en la penumbra isquémica. Las terapias de reperfusión incluyen la trombolisis intravenosa, la trombolisis intraarterial y la trombectomía mecánica endovascular. Las guías integrales para el manejo temprano del ictus isquémico agudo se actualizaron en 2013; este capítulo solo puede resumir brevemente algunas de estas importantes guías. A lo largo de esta sección, las recomendaciones que no se citan directamente provienen de este documento; cualquier desviación de estas guías se indicará explícitamente. La técnica más eficaz para mejorar la recuperación del ictus embólico es la pronta restauración de la perfusión espontánea lo antes posible tras el inicio del ictus. Hasta la fecha, a pesar de la evidencia reciente de que, para la oclusión de grandes vasos, la trombectomía mecánica intracraneal es superior a la intervención farmacológica sola, el único método aprobado por la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) es el uso del agente trombolítico activador tisular del plasminógeno recombinante (rtPA). Como era de esperar, los efectos del rtPA empeorarían notablemente el ictus hemorrágico. Por lo tanto, detectar, clasificar y tratar el ictus rápidamente tras su aparición es fundamental para un resultado favorable. Los trombolíticos no pueden utilizarse en pacientes con alto riesgo de hemorragia, como aquellos con traumatismo craneoencefálico, cirugía reciente o con disminución de la capacidad de coagulación debido a fármacos como la warfarina, los inhibidores directos de la trombina y los inhibidores directos del factor Xa. Los pacientes tratados con warfarina con un INR inferior a 1,4 pueden recibir tratamiento con rtPA; sin embargo, los nuevos fármacos presentan un problema, ya que resulta difícil evaluar la función de coagulación en pacientes que los toman, y las recomendaciones al respecto están en revisión.
El principal efecto secundario del rtPA es la hemorragia intracerebral, que puede ser devastadora. Es fundamental realizar y analizar una tomografía computarizada sin contraste poco después de la llegada del paciente al hospital para descartar un ictus hemorrágico, ya que el rtPA debe administrarse dentro de las 3 horas posteriores al inicio de un ictus oclusivo para que sea efectivo. Las guías de la AHA/ASA indican que el rtPA puede administrarse a pacientes entre 3 y 4,5 horas después del inicio del ictus si son menores de 80 años, no toman anticoagulantes orales y no tienen antecedentes de ictus ni diabetes; esta recomendación no es una indicación de la FDA para el rtPA. Es evidente que cuanto antes se administre el rtPA, mejor será el pronóstico.
Al explorar terapias adicionales, es fundamental identificar a los pacientes en quienes un área de perfusión reducida no ha progresado a daño neuronal irreversible. Estos estudios utilizan técnicas de imagen avanzadas para identificar el tejido en riesgo (penumbra) que aún puede salvarse si se restablece la reperfusión. La resonancia magnética ponderada por difusión (DWI) identifica las áreas isquémicas centrales donde el agua se ha desplazado al compartimento intracelular y ha reducido su difusividad. Las áreas que aún no se han convertido en isquemia central pueden potencialmente salvarse del daño irreversible. La resonancia magnética ponderada por perfusión (PMPR) indica regiones con perfusión reducida que, en última instancia, progresarán a daño irreversible si no se reperfunden. La relación entre el volumen de tejido isquémico penumbral y el volumen del núcleo infartado se denomina discrepancia penumbral. Sin embargo, debido a la importancia de una reperfusión rápida y al retraso en la realización de la resonancia magnética, la práctica actual utiliza la angiotomografía computarizada multifásica para determinar si un vaso grande está ocluido, la ubicación de la oclusión y una evaluación del volumen del núcleo del infarto o la discrepancia en la penumbra. Dado que el rtPA es menos eficaz y las técnicas de eliminación mecánica intraarterial de coágulos son más eficaces para oclusiones grandes, debe considerarse la trombectomía mecánica con rapidez; con todas las técnicas de reperfusión, un aumento en el tiempo de reperfusión equivale a una mayor pérdida de tejido cerebral. Una región con perfusión reducida que aún no ha progresado a un daño irreversible se beneficiará de la terapia de reperfusión endovascular.
El Merci Retriever (Concentric Medical, Inc., Mountain View, CA), el sistema Penumbra (Penumbra, Inc., Alameda, CA), el dispositivo Solitaire (Covidien, Irvine, CA) y el Trevo Retriever (Stryker Neurovascular, Freemont, CA) están aprobados por la Administración de Alimentos y Medicamentos de EE. UU. para la extracción mecánica de coágulos. El Merci Retriever, un dispositivo con forma de sacacorchos, ha demostrado proporcionar una mayor tasa de buenos resultados clínicos en comparación con los controles históricos, aunque la tasa de hemorragia intracerebral fue del 7,8 %, similar a la del rt-PA intravenoso. Estudios recientes han demostrado que los recuperadores de stent Solitaire FR y Trevo son superiores al dispositivo Merci. En todos los casos, los pacientes candidatos a rt-PA intravenoso deben recibir dicho tratamiento lo antes posible, incluso si se está considerando la extracción mecánica intraarterial del coágulo. Estudios recientes han encontrado un claro beneficio en los resultados del tratamiento mecánico intraarterial para el ictus. La experiencia, la rapidez del tratamiento y los nuevos dispositivos han mejorado esta técnica hasta el punto de que su disponibilidad a demanda se está convirtiendo rápidamente en el estándar de atención para los centros de ictus. La actualización de la guía de la AHA/ASA de 2015 concluye que «se ha demostrado que ciertos procedimientos endovasculares proporcionan un beneficio clínico en pacientes seleccionados con ictus isquémico agudo. Los sistemas de atención deben organizarse para facilitar la prestación de esta atención». Los pacientes candidatos a trombectomía mecánica deben tener oclusiones confirmadas en la circulación intracraneal anterior proximal y ausencia de grandes lesiones isquémicas centrales. Sin embargo, aún no existe consenso sobre el tipo de neuroimagen ni sobre los criterios exactos de imagen para la elegibilidad del paciente; la rapidez de la evaluación es fundamental, ya que los retrasos conllevan un mayor infarto cerebral. El intervalo de tiempo aceptado para el tratamiento con trombectomía mecánica es de 6 horas desde el inicio de los síntomas. Por ahora, el tratamiento eficaz más utilizado para el ictus isquémico sigue siendo la administración de rtPA intravenoso dentro de las 3 horas posteriores al inicio del ictus, o hasta 4,5 horas en ciertos pacientes. Este tratamiento se utiliza poco debido al corto margen de tiempo para la administración segura de rtPA, las numerosas contraindicaciones para su uso y la frecuente demora de los pacientes en llegar al hospital tras la aparición de los síntomas. Este problema puede abordarse, y de hecho se está abordando, mediante la educación comunitaria sobre los signos del ictus a través de la campaña FAST (Cara, Brazos, Habla, Tiempo para llamar al 911) y la necesidad de traslado de urgencia a un hospital adecuado con un centro especializado en ictus. Las guías para el manejo y tratamiento precoz del ictus se actualizan con frecuencia y deben consultarse para obtener las recomendaciones más recientes.
Hipotermia
La hipotermia profunda se ha utilizado durante mucho tiempo en la cirugía cardíaca neonatal para proteger contra el daño cerebral irreversible durante el paro circulatorio. También se ha utilizado en la reparación de aneurismas gigantes. Sin embargo, la hipotermia profunda (27 °C o menos) presenta numerosas complicaciones que limitan su utilidad (Recuadro 3). La hipotermia profunda reduce el metabolismo cerebral de tal manera que el cerebro puede sobrevivir periodos relativamente largos sin perfusión (Recuadro 4). Estudios experimentales indican que la hipotermia moderada tiene un efecto protector sin muchas de las complicaciones de la hipotermia profunda, aunque se ha documentado depresión miocárdica. Existen numerosos estudios in vitro e in vivo en animales que respaldan el uso de la hipotermia moderada para proteger contra el daño isquémico. De hecho, la hipotermia moderada se ha generalizado a pesar de que no se ha demostrado de forma inequívoca que mejore la recuperación en un ensayo clínico importante. Un estudio europeo publicado en 2002 indica que la hipotermia leve, con una temperatura objetivo de 32 °C a 34 °C, tras un paro cardíaco mejora el pronóstico neurológico y la supervivencia a los 6 meses. Sin embargo, estudios más recientes y de mayor envergadura no han encontrado beneficios en la hipotermia leve en comparación con mantener la temperatura por debajo de 36 °C; evitar la hipertermia parece ser importante y podría explicar el beneficio observado en estudios anteriores. La recomendación de hipotermia tras un paro cardíaco podría ser reemplazada pronto por la de evitar la hipertermia. No obstante, con respecto a la hipotermia tras un ictus, no existe evidencia de beneficio de Clase I; el enfriamiento a niveles entre 34 °C y 35 °C reduce las complicaciones y el recalentamiento lento es importante para minimizar los efectos adversos.36 La hipotermia leve no mejoró el resultado de la cirugía de aneurisma intracraneal y una revisión sistemática Cochrane no halló beneficios ni perjuicios derivados de la hipotermia durante un ictus agudo. Si la hipotermia demuestra algún beneficio para ciertos tipos de pacientes con ictus, será necesario determinar con mayor precisión su grado y duración, así como la velocidad de recalentamiento. Es evidente que incluso cantidades leves de hipertermia empeoran el pronóstico clínico de la isquemia y aumentan el daño neuronal, y esto debe evitarse con sumo cuidado.
- Depresión miocárdica
- Disritmia, incluyendo fibrilación ventricular
- Hipotensión
- Perfusión tisular inadecuada
- Isquemia
- Trombocitopenia
- Fibrinólisis
- Disfunción plaquetaria
- Aumento del sangrado
- Metabolismo enlentecido de los agentes anestésicos
- Bloqueo neuromuscular prolongado
- Aumento del catabolismo proteico
- Aumento del consumo de oxígeno
- Aumento de la producción de dióxido de carbono
- Aumento del gasto cardíaco
- Desaturación arterial de oxígeno
- Inestabilidad hemodinámica
- Reducción del metabolismo cerebral de oxígeno (aproximadamente un 6% a 7% por cada 1°C de disminución de la temperatura central)
- Reducción de la tasa de utilización de glucosa
- Disminución de la acumulación de glutamato y glicina (neurotransmisores excitatorios)
- Supresión de la producción de radicales libres de oxígeno, peróxido de hidrógeno y óxido nítrico
- Atenuación de la cascada apoptótica (muerte celular programada)
- Disminución de la producción de citocinas proinflamatorias (IL-1, IL-6, TNF-α)
- Preservación de la integridad de la barrera hematoencefálica (reducción del edema cerebral)
- Reducción de la respuesta astrocítica y microglial
- Supresión de la actividad convulsiva detectable por electroencefalograma (EEG) e inhibición de descargas epileptiformes
Glucosa
La glucosa es la principal fuente de energía para las neuronas del cerebro, y algunos estudios in vitro informaron una mejor recuperación con hiperglucemia. Sin embargo, estudios in vivo y clínicos encontraron un claro empeoramiento del daño con hiperglucemia, lo que se cree que se debe a una acidosis celular aumentada. Se desconoce el mecanismo preciso por el cual la hiperglucemia exacerba el daño. Las recomendaciones clínicas son mantener niveles normales de glucosa en suero y tratar la hiperglucemia mayor de 180 mg/dL para reducir el valor de glucosa a valores más cercanos a la normalidad. Es importante que el paciente no tenga hipoglucemia, ya que la hipoglucemia también empeoraría el pronóstico. El control estricto e intensivo de la glucosa no mejoró el pronóstico después de un accidente cerebrovascular, y un estudio más reciente demostró una mayor mortalidad en pacientes controlados con niveles de 81 a 108 mg por decilitro en comparación con pacientes controlados con un objetivo de 140-180 mg por decilitro. Los episodios de hipoglucemia debido al uso excesivo de insulina para controlar los niveles de glucosa probablemente expliquen el peor pronóstico con el control intensivo de la glucosa. Se recomienda controlar la glucosa para mantenerla por debajo de 180, pero un control estricto es claramente perjudicial; existen pruebas más sólidas que indican la necesidad de tratar la hipoglucemia si la glucosa cae por debajo de 60 mg/dL.
Agentes farmacológicos
Estudios en animales han demostrado que varios agentes pueden mejorar el resultado de un accidente cerebrovascular experimental; sin embargo, existe controversia sobre por qué ninguno de estos agentes ha producido una mejoría clínica. Se han propuesto muchos fármacos como posibles agentes para reducir el daño neuronal permanente posterior a la isquemia, pero ninguno ha demostrado ser útil en ensayos clínicos. La base teórica para elegir fármacos que bloqueen vías dañinas específicas es sólida. Sin embargo, bloquear una sola vía de daño puede no ser eficaz, debido a las múltiples vías paralelas que conducen al daño permanente (véase la figura 7). Por ejemplo, se pueden bloquear los canales de calcio sensibles al voltaje, pero el calcio citoplasmático puede aumentar mediante la entrada a través del canal iónico del receptor NMDA o la liberación desde orgánulos intracelulares. Por lo tanto, una terapia eficaz podría requerir múltiples agentes para bloquear las vías paralelas simultáneamente.
Es importante reconocer que ningún agente farmacológico ni combinación de agentes que muestre una protección clara en animales ha demostrado mejorar la recuperación neurológica clínicamente tras un ictus. Sin embargo, algunos agentes parecen prometedores en estudios con animales y podrían resultar eficaces en la práctica clínica. Un problema importante en los tratamientos del ictus, y una de las razones de la discrepancia entre los resultados en animales y en humanos, es que la mayoría de los estudios con animales aplican los agentes protectores antes o durante el evento isquémico, mientras que el tratamiento clínico del ictus siempre se retrasa. En el entorno perioperatorio, los fármacos y tratamientos pueden aplicarse antes del evento isquémico, al inicio de una cirugía de alto riesgo; por lo tanto, los agentes que no protegen contra el ictus cuando se usan después del evento pueden ser eficaces si se administran antes de la cirugía. Dado que muy pocos pacientes sometidos a cirugía de alto riesgo sufrirán un evento isquémico, los agentes utilizados deben tener un alto factor de seguridad y/o ser necesarios para la cirugía (por ejemplo, los anestésicos). Los efectos nocivos de cualquier agente utilizado se compartirán entre todos los pacientes, pero sus efectos protectores solo beneficiarán a los pacientes con ictus isquémico. En las siguientes secciones se analizan las clases de estrategias potencialmente protectoras.
Bloqueo de sodio
Se ha demostrado que bloquear la entrada de sodio durante la anoxia y la isquemia mejora la recuperación. La despolarización neuronal durante la anoxia y la isquemia provoca un flujo masivo de sodio y calcio hacia el interior de las neuronas y de potasio hacia el exterior. El bloqueo de la entrada de sodio retrasa y atenúa la despolarización y la disminución de ATP durante la anoxia y la isquemia. La lidocaína ha mejorado la recuperación al retrasar y atenuar la despolarización anóxica/isquémica y reducir la entrada de sodio anóxica cuando se administra en concentraciones que no bloquean los canales de sodio en condiciones normales. La lidocaína redujo el tamaño del infarto y mejoró el pronóstico neurológico tras una isquemia cerebral focal. Parece actuar, al menos en parte, bloqueando las vías apoptóticas en la penumbra. Cuando la aplicación de lidocaína se retrasó hasta 45 minutos después del inicio de la isquemia cerebral focal, se observó una mayor supervivencia neuronal en el núcleo y la penumbra, pero el tamaño del infarto no se redujo significativamente. Esto ilustra la importancia de la administración rápida de los fármacos para que tengan algún efecto beneficioso. La administración de una dosis antiarrítmica de lidocaína mejoró la supervivencia de las neuronas piramidales CA1 de ratas tras una isquemia cerebral global transitoria, así como el rendimiento en una tarea cognitiva dependiente del hipocampo. Dos pequeños estudios clínicos sobre lidocaína han indicado una mejoría en la función cognitiva tras la cirugía cardíaca. Un estudio clínico más reciente no halló mejoría con la administración de lidocaína durante la cirugía cardíaca; sin embargo, un subgrupo, pacientes no diabéticos, sí mostró mejoría. Un estudio que examina la lidocaína en pacientes no diabéticos sometidos a cirugía cardíaca está en curso. Un estudio reciente con lidocaína administrada tras una cirugía supratentorial no halló mejoría, aunque el daño en el grupo no tratado fue muy bajo a los 6 meses. La recuperación fue, sin embargo, similar a la de pacientes no neuroquirúrgicos con características demográficas similares. Los futuros estudios sobre lidocaína deberían examinar a pacientes de mayor edad y/o con enfermedades más graves, con una mayor probabilidad de disfunción cognitiva en el grupo no tratado.
Bloqueo de calcio
Se ha demostrado que la nimodipina, un bloqueador del canal de calcio dependiente de voltaje, mejora la recuperación de la hemorragia subaracnoidea, mientras que la nicardipina, un fármaco estrechamente relacionado, no lo hace; por lo tanto, la nimodipina solo está indicada en este contexto para prevenir o tratar el vasoespasmo. Un amplio estudio clínico sobre la eficacia de la nimodipina tras un ictus se interrumpió debido a una mayor mortalidad en el grupo tratado con nimodipina. Es evidente que la nimodipina no se recomienda tras una isquemia cerebral. De hecho, durante la isquemia y la anoxia, los canales de calcio ya están inhibidos y no se observó protección directa de las neuronas con nimodipina en preparaciones in vitro. El magnesio, un agente que bloquea muchos canales dependientes de voltaje y activados por neurotransmisores (incluidos los canales activados por NMDA que inducen excitotoxicidad), reduciría la entrada de calcio y otros iones. Recientemente se ha demostrado que el magnesio es beneficioso durante la isquemia cerebral focal; sin embargo, un ensayo clínico no mostró beneficios con la administración intravenosa de magnesio tras un ictus. Hubo un subgrupo de ictus lacunar que sí mostró beneficio con el magnesio, pero esto requeriría un nuevo ensayo clínico a gran escala para confirmarlo. Un estudio sobre el uso de magnesio en partos prematuros para proteger el cerebro infantil del daño no mostró una mejoría significativa con el tratamiento con magnesio. Claramente, se necesitan más estudios antes de poder recomendarlo. Un problema importante es el acceso limitado del magnesio al sistema nervioso central debido a su escasa permeabilidad a través de la barrera hematoencefálica. Una crítica a los ensayos clínicos con magnesio, y a muchos otros estudios, fue el retraso en la administración de los fármacos; en la mayoría de los estudios con animales, los fármacos se administraron antes o muy poco después del inicio de la isquemia, lo cual no es aplicable clínicamente. Al momento de redactar este informe, se acaba de completar un estudio que administraba magnesio en la ambulancia; si bien demostró que la administración rápida de magnesio después de un ictus era posible, no se observó ningún beneficio. El bloqueo de las vías secundarias activadas por calcio durante y después de la isquemia parece prometedor en estudios con animales.
Eliminación de radicales libres
Los radicales libres se han relacionado con el daño celular y, posteriormente, con el daño neuronal tras la isquemia. Se cree que tanto el daño apoptótico como el necrótico tienen un componente de daño por radicales libres. Se ha demostrado que el uso de captadores de radicales libres como NXY-059, alfa-tocoferol, tirilizad y N-terc-alfa-fenil-butilnitrona (PBN) mejora la isquemia en animales; sin embargo, ninguno de estos agentes ha demostrado mejorar el pronóstico clínico. Los fármacos antiinflamatorios como la metilprednisolona tampoco han demostrado mejorar la recuperación tras un traumatismo cortical o isquemia. Los corticosteroides suprimen el sistema inmunitario, aumentan las infecciones y pueden, de hecho, potenciar el daño causado por los radicales libres. Se ha relacionado el óxido nítrico con el aumento del daño neuronal, y el lubeluzol, un inhibidor de su formación, ha mostrado cierto potencial en estudios con animales, pero no se ha encontrado que beneficie a los pacientes con isquemia.
Agentes que reducen la excitotoxicidad
Los aminoácidos excitatorios están implicados en la cascada de daño que se produce tras la isquemia, el traumatismo y la epilepsia. Si bien los bloqueadores de los receptores de glutamato NMDA y AMPA han mejorado la recuperación in vitro e in vivo en diversas preparaciones, los resultados de los ensayos clínicos han sido decepcionantes. Al parecer, estos agentes son tóxicos y pueden causar daño neuronal. De hecho, algunos ensayos clínicos con estos agentes se han interrumpido prematuramente debido a efectos adversos.
Una alternativa al bloqueo de la actividad excitatoria es potenciar la actividad inhibitoria, lo que también reduciría la excitotoxicidad. El clometiazol y el diazepam, dos potenciadores del GABA, no mejoraron el pronóstico tras un ictus.
Agentes antiapoptóticos
Los estudios sobre la apoptosis indican que los bloqueadores específicos de las caspasas y los moduladores de la apoptosis podrían mejorar la recuperación tras isquemia, traumatismos o epilepsia. Si bien los experimentos con animales in vivo son prometedores, no se ha demostrado que estos agentes mejoren el pronóstico clínico. Una técnica más útil podría consistir en estimular a las neuronas para que sinteticen proteínas antiapoptóticas, como Bcl-2 y Bcl-xl, mediante el preacondicionamiento con ciertos anestésicos volátiles.
Citocinas y factores tróficos
Las citocinas como el factor de necrosis tumoral alfa y la interleucina-1β pueden activar el sistema inmunitario y aumentar el daño; de hecho, se ha demostrado que los anticuerpos contra estos compuestos reducen el daño isquémico cerebral en algunos animales. Sin embargo, el factor de necrosis tumoral alfa también puede ser beneficioso, favoreciendo la supervivencia neuronal en ciertas circunstancias; por lo que su inhibición puede tener resultados mixtos.
Las neuronas poseen receptores para factores tróficos como el factor de crecimiento nervioso, las neurotrofinas y el factor de crecimiento derivado del cerebro, esenciales para la supervivencia neuronal incluso en ausencia de lesión. Estos factores activan receptores que fosforilan aminoácidos en ciertas proteínas, inhibiendo así la apoptosis. Si estos factores de crecimiento no están presentes, los receptores no se activan y las proteínas no se fosforilan; en consecuencia, las neuronas sufren apoptosis. La pérdida de factores de crecimiento tras la degeneración neuronal por isquemia puede agravar la pérdida neuronal tardía.
La eritropoyetina es un factor trófico para las células sanguíneas que también está presente en el sistema nervioso. Estudios en animales indican que podría proteger a las neuronas de la apoptosis tras la isquemia mediante la activación de la vía antiapoptótica del factor trófico. A pesar de las investigaciones en curso, no existe evidencia concluyente de beneficio clínico con la eritropoyetina.
Agentes anestésicos
Se han examinado los agentes anestésicos por su capacidad para mejorar la recuperación de la isquemia. La teoría intuitiva es que reducen la actividad neuronal y la tasa metabólica y, por lo tanto, deberían disminuir la demanda de energía, aumentar el suministro de energía y atenuar el daño isquémico (Tabla 1). Sin embargo, los diferentes anestésicos también tienen acciones específicas, incluyendo efectos sobre las vías de señalización intracelular, las conductancias iónicas y los neurotransmisores, así como efectos hemodinámicos sistémicos y cerebrales; estas otras acciones pueden ayudar a explicar sus efectos diferenciales sobre el daño neuronal (Tabla 2). Los estudios que comparan la anestesia general con la sedación consciente para el tratamiento endovascular del ictus agudo indican que los pacientes con anestesia general tienen un peor pronóstico. El mecanismo no está claro, pero esto debería llevar a la cautela al concluir que los anestésicos pueden mejorar el pronóstico en pacientes como lo hacen en animales. Es posible que una reducción de la presión arterial debido a la anestesia general esté impulsando el peor pronóstico independientemente de los efectos anestésicos directos. Dado que todos los estudios sobre el tipo de anestesia en el tratamiento endovascular del ictus agudo son retrospectivos, se planteó la salvedad de que los pacientes que requerían anestesia general probablemente habían sufrido un ictus más grave y presentaban un peor estado neurológico preoperatorio. Un estudio reciente de 369 pacientes con ictus procedentes de un registro holandés, que recibieron o no anestesia general (siguiendo estrictamente el protocolo del hospital local), mostró mejores resultados sin anestesia general, pero sin desequilibrio en la gravedad del ictus entre los grupos. La causa de esta diferencia es hipotética, pero el tratamiento se inició antes en los pacientes no anestesiados. Aunque los anestésicos reducen el daño cuando se administran antes de la isquemia, en el contexto de un ictus es improbable que alcancen la zona de beneficio debido a la reducción del flujo sanguíneo y la administración del fármaco al tejido que más necesita protección.
| Anestésico | FSC | CMRO2 | Vasodilatación Cerebral Directa |
|---|---|---|---|
| Halotano | ↑ ↑ ↑ | ↓ | Sí |
| Enflurano | ↑ ↑ | ↓ | Sí |
| Isoflurano | ↑ | ↓ ↓ | Sí |
| Desflurano | ↑ | ↓ ↓ | Sí |
| Sevoflurano | ↑ | ↓ ↓ | Sí |
| N2O | ↑ | ↑ | — |
| N2O con anestésicos volátiles | ↑ ↑ | ↑ | — |
| N2O con anestésicos intravenosos | 0 | 0 | — |
| Tiopental | ↓ ↓ ↓ | ↓ ↓ ↓ | No |
| Etomidato | ↓ ↓ | ↓ ↓ | No |
| Propofol | ↓ ↓ | ↓ ↓ | No |
| Midazolam | ↓ | ↓ | No |
| Dexmedetomidina | ↓ | 0 | No |
| Ketamina | ↑ ↑ | ↑ | No |
| Fentanilo | ↓/0 | ↓/0 | No |
| Agente | Protege la Respuesta Electrofisiológica | Retrasa la Despolarización Hipóxica | Reduce el Ingreso de Na+ | Mejora el Trifosfato de Adenosina | Reduce el Ingreso de Ca2+ |
|---|---|---|---|---|---|
| Tiopental (600 µM) | Sí | Sí | Sí | Sí | Sí |
| Midazolam (100 µM) | Sí | — | — | Sí | Sí |
| Propofol (20 µg/mL) | No | No | Sí | Sí | Sí |
| Etomidato (3 µg/mL) | No | — | No | No | — |
| Lidocaína (10 µM) | Sí | Sí | Sí | Sí | No |
| Lidocaína (100 µM) | Sí | Sí | Sí | Sí | Sí |
| Óxido nitroso (50%) | No | — | No | No | No |
| Isoflurano (2%) | No | No | Sí | Sí | No |
| Sevoflurano (4%) | Sí | Sí | Sí | Sí | Sí |
| Desflurano (6%) | Sí | Sí | Sí | Sí | Sí |
Barbitúricos
Los barbitúricos son los únicos anestésicos que han demostrado eficacia protectora clínicamente, pero solo en un contexto muy específico. El mecanismo de su protección no se ha establecido y, de hecho, podría ser multifacético. Entre sus múltiples acciones, el tiopental bloquea los flujos de Na, K y calcio, neutraliza los radicales libres, previene las convulsiones, mejora el flujo sanguíneo regional y disminuye la presión intracraneal (PIC). Quizás sean las múltiples acciones que bloquean vías dañinas paralelas las que permiten que este agente proteja contra el daño isquémico. Es importante destacar que se necesitaron dosis muy altas de barbitúricos para inducir un coma y demostrar una mejoría clínica tras la cirugía cardíaca. Los estudios in vitro también mostraron una mayor eficacia con dosis muy altas.
Etomidato
El etomidato, al igual que el tiopental, reduce la tasa metabólica cerebral cuando se administra en dosis de supresión de descargas; sin embargo, no comparte muchas de las otras acciones del tiopental. No se ha demostrado que el etomidato mejore la recuperación del daño isquémico y anóxico en condiciones normales. No se puede asumir que un anestésico que reduce la tasa metabólica cerebral en la misma medida que el tiopental proporcionará la misma protección cerebral. Estudios en animales encontraron que el etomidato inhibe la NO sintasa, lo que podría reducir la perfusión del tejido cerebral, y el etomidato empeoró el pronóstico de la isquemia cerebral focal en comparación con el halotano. Estos estudios sugieren que el etomidato no es el fármaco de elección si se anticipa isquemia cerebral.
Propofol
El propofol es un anestésico intravenoso de uso extendido. Estudios en animales indican que puede reducir el daño isquémico, aunque su potencia podría ser menor que la del tiopental. El propofol demostró una protección similar a la del pentobarbital y resultó beneficioso en comparación con animales conscientes. Al igual que con otros agentes anestésicos, no se ha demostrado su eficacia protectora clínica.
Dexmedetomidina
La dexmedetomidina es un agonista adrenérgico alfa 2a con efectos sedantes, analgésicos y ansiolíticos. Reduce la actividad simpática al inhibir la liberación de noradrenalina (norepinefrina) desde las terminaciones nerviosas presinápticas. La dexmedetomidina puede causar hipotensión, lo cual podría ser perjudicial en pacientes con isquemia cerebral. Sin embargo, estudios en animales que examinaron la lesión isquémica del cerebro y la médula espinal demostraron una reducción de la lesión con dexmedetomidina, lo que indica un efecto protector directo del fármaco. No se han realizado estudios que demuestren eficacia protectora clínica.
Xenón
El xenón es un gas inerte que presenta efectos anestésicos a dosis muy altas; no se utiliza clínicamente como anestésico, pero ha demostrado eficacia protectora en animales adultos. Se ha demostrado que dosis subanestésicas de xenón reducen el daño cerebral tras la asfixia neonatal y pueden atenuar el deterioro de la memoria inducido por la anestesia en ratones neonatos. El xenón no se utiliza clínicamente.
Óxido nitroso
En estudios con animales, se ha demostrado que el óxido nitroso reduce la recuperación de la isquemia y la anoxia en comparación con otros anestésicos; por lo tanto, se requiere precaución al usarlo en pacientes con potencial de perfusión cerebral comprometida.
Benzodiazepinas
La benzodiazepina más utilizada en anestesia es el midazolam, y se ha estudiado su efecto sobre el metabolismo cerebral y el daño isquémico. Las benzodiazepinas potencian la inhibición neuronal en el sistema nervioso y reducen el metabolismo cerebral al aumentar el efecto del neurotransmisor ácido gamma-aminobutírico (GABA) sobre el receptor GABA. Se ha demostrado que dosis altas de midazolam reducen el metabolismo cerebral y el flujo sanguíneo cerebral, efecto que se revierte con el antagonista de las benzodiazepinas flumazenil. El midazolam mejoró la recuperación neuronal tras anoxia e isquemia en animales, pero no existen estudios que demuestren un mejor resultado clínico. El flumazenil debe utilizarse con precaución, o incluso evitarse, para revertir los efectos de las benzodiazepinas en situaciones en las que un aumento de la tasa metabólica cerebral sea indeseable, ya que se ha demostrado que este fármaco aumenta la tasa metabólica cerebral, el flujo sanguíneo cerebral y la presión intracraneal (PIC).
Agentes anestésicos volátiles
El isoflurano es un anestésico volátil cuya eficacia protectora es controvertida. No causa mayor daño y parece tener un mejor resultado que la anestesia con fentanilo y óxido nitroso. El sevoflurano y el desflurano tienen efectos metabólicos y sobre el flujo sanguíneo similares a los del isoflurano, y se ha informado que tienen efectos neuroprotectores (véase el recuadro 1). Hay indicios de que el isoflurano, pero no el sevoflurano, aumenta los niveles de Ca citosólico y puede ser citotóxico para las neuronas en cultivos celulares. Se observó que el sevoflurano mejoraba la recuperación en cortes cerebrales; retrasó y atenuó la despolarización hipóxica/isquémica y redujo los aumentos de Ca y Na dentro de las neuronas. A concentraciones alveolares mínimas equivalentes, el sevoflurano fue más eficaz que el isoflurano y demostró una mejoría sostenida tras la isquemia cerebral en comparación con la anestesia con óxido nitroso y fentanilo. El desflurano también demostró ser protector en cortes cerebrales y tras isquemia cerebral in vivo.
Preacondicionamiento
Tanto en el tejido cardíaco como en el cerebral, el preacondicionamiento isquémico —un breve período de isquemia que permite la recuperación— puede aumentar la resistencia del tejido a un período de isquemia más prolongado y normalmente dañino. Sin embargo, el preacondicionamiento isquémico puede provocar daños sutiles. Se ha demostrado que los anestésicos, administrados antes de la isquemia, inducen preacondicionamiento; es probable que estos agentes sean menos dañinos que el preacondicionamiento isquémico. Existe abundante evidencia que indica que el isoflurano mejora la recuperación de la isquemia cerebral mediante el preacondicionamiento de las neuronas, pero la mayoría de los estudios se han realizado en animales machos. Un estudio posterior indica que los ratones machos, pero no las hembras, presentan una mejor recuperación del preacondicionamiento inducido por isoflurano administrado un día antes de la isquemia. Por lo tanto, es importante recordar que puede haber aspectos específicos de género en la protección contra el daño cerebral isquémico. Este es un tema que requiere mayor investigación.
Tanto el preacondicionamiento anestésico como el isquémico presentan dos cronologías: el preacondicionamiento retardado se manifiesta un día después del estímulo de preacondicionamiento y dura varios días; el preacondicionamiento inmediato requiere tratamiento solo entre minutos y una hora antes de la isquemia. Se ha demostrado que el sevoflurano induce preacondicionamiento in vitro e in vivo si se administra poco antes de la isquemia. El mecanismo y el alcance de la protección con sevoflurano in vitro fueron similares a los observados cuando el sevoflurano se administró antes y durante la hipoxia; este hallazgo sugiere que una parte importante de la protección inducida por el sevoflurano se debe a una alteración de las vías bioquímicas antes de la lesión. El preacondicionamiento con sevoflurano durante 60 minutos, comenzando 90 minutos antes de la isquemia, con un 4 % o un 2 % de sevoflurano, aumentó el número de células piramidales CA1 supervivientes 6 semanas después de una isquemia cerebral global transitoria en ratas (Fig. 8).
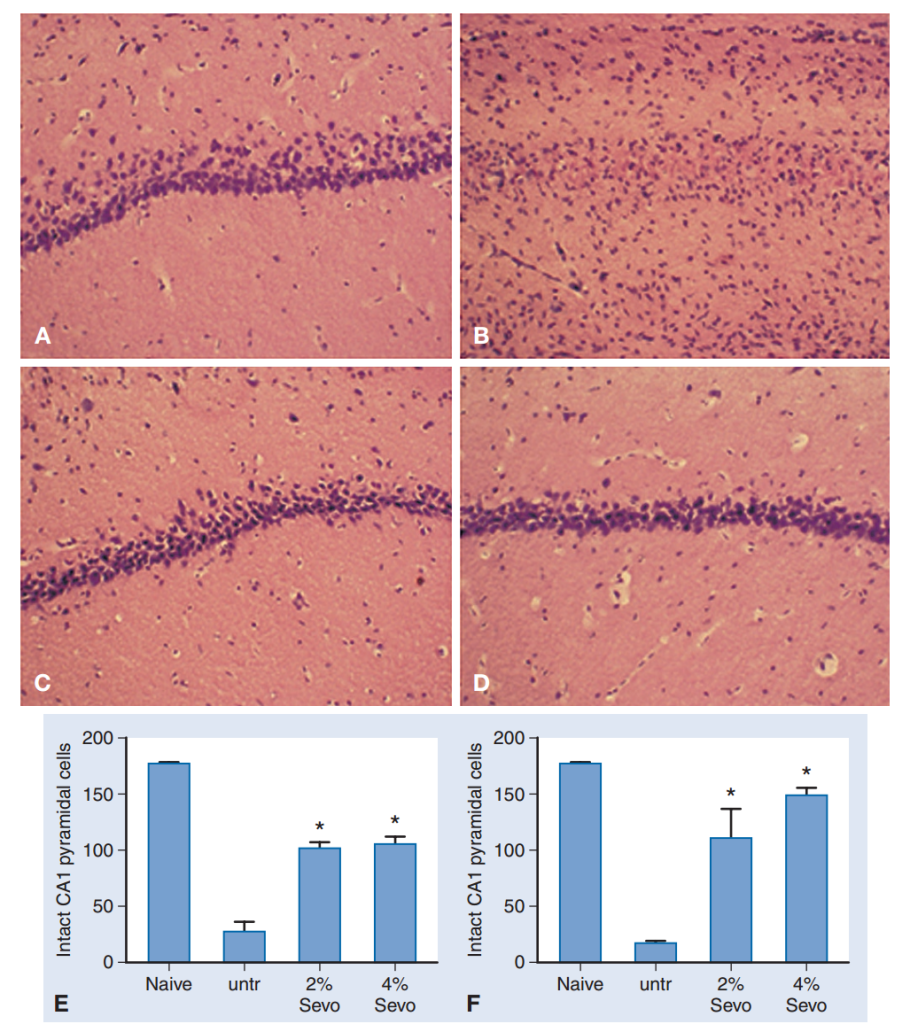
El preacondicionamiento anestésico debe demostrarse clínicamente antes de que pueda aplicarse de forma generalizada; sin embargo, si se elige un anestésico para un paciente con riesgo de isquemia, podría ser prudente utilizar un agente que haya demostrado ser beneficioso en animales.
Agentes anestésicos en niños pequeños
Estudios en animales han demostrado que los agentes anestésicos, administrados a animales neonatos, pueden provocar déficits cognitivos y conductuales. La interacción social en ratones tratados con sevoflurano durante el período neonatal se reduce, lo que confirma que la función conductual se ve alterada por la exposición a la anestesia. Estas observaciones indican que podrían ser necesarios estudios clínicos que examinen los déficits conductuales en niños tras la exposición a la anestesia. Existen indicios de que la anestesia puede tener efectos perjudiciales en niños pequeños, aunque resulta difícil separar los efectos directos del anestésico de las condiciones quirúrgicas y médicas que motivaron la cirugía y, por lo tanto, la anestesia. Queda por determinar si los anestésicos, y cuáles y en qué dosis, provocan déficits cognitivos en niños. Las ventajas teóricas de posponer la cirugía electiva deben sopesarse frente a los posibles daños que conlleva un retraso en la intervención.
Tratamiento
En resumen, los anestésicos tienen efectos diferenciales sobre el metabolismo neuronal, los flujos iónicos y los potenciales de membrana. Su mecanismo de acción es múltiple, lo que complica los estudios científicos, pero puede mejorar la protección clínica. Se ha demostrado en estudios con animales que la hipotermia, la lidocaína, el tiopental y el sevoflurano protegen contra la isquemia.
Clínicamente, la hipotermia y la lidocaína son las opciones más prometedoras, pero no existe evidencia concluyente de que mejoren la recuperación de la isquemia en pacientes. El tiopental requiere concentraciones muy altas para ejercer su efecto protector, mientras que el sevoflurano necesita más estudios para determinar si protege a los pacientes cuando se administra en concentraciones clínicamente útiles. Dado que la anestesia es necesaria durante la cirugía, podría ser prudente elegir un anestésico que parezca protector en animales, incluso si no se ha demostrado su protección clínicamente; el sevoflurano podría ser una buena opción, ya que parece ser protector en el rango de dosis clínicas. El tiopental requiere una dosis demasiado alta para usarse como anestésico en estos casos, ya que retrasaría el despertar; sin embargo, su uso en cuidados intensivos, donde el despertar no es un problema, podría ser beneficioso. Limitar la isquemia mediante la mejora de la perfusión es el mecanismo más eficaz para prevenir el daño neuronal causado por el ictus. La trombolisis y la prevención de la formación de coágulos y la embolización son estrategias eficaces.
Daños Provocados Por Convulsiones
La actividad epiléptica (estado epiléptico) consiste en descargas repentinas, excesivas y sincrónicas de un gran número de neuronas. Además de en pacientes con epilepsia establecida, este aumento masivo de actividad puede observarse en pacientes con desequilibrios iónicos y electrolíticos, trastornos del metabolismo cerebral, infecciones, tumores cerebrales, traumatismos craneoencefálicos y fiebre. El electroencefalograma muestra espigas, que son cambios rápidos de voltaje que corresponden a una actividad excesiva en muchas neuronas. Durante la actividad epileptiforme, los iones de sodio y calcio entran en las células y el potasio sale. Por lo tanto, las células utilizan más energía (ATP) para el bombeo de iones. El elevado nivel de potasio extracelular resultante puede ser responsable de la gran y progresiva despolarización neuronal que se observa comúnmente. Los mecanismos que conducen al daño neuronal permanente en la epilepsia pueden ser similares a los que dañan las células durante la isquemia. La actividad epiléptica provoca la liberación de glutamato, la activación del receptor NMDA y la excitotoxicidad; esto puede causar daño neuronal. La activación de los receptores metabotrópicos de glutamato puede contribuir a la excitabilidad excesiva y prolongar las convulsiones. Los niveles de calcio intracelular aumentan, lo que posiblemente precipiten el daño. Existe evidencia de que al menos parte del daño neuronal permanente es de tipo apoptótico. Es evidente que durante la actividad epileptiforme la demanda de energía y, por lo tanto, la tasa metabólica cerebral y el flujo sanguíneo aumentan considerablemente. Por consiguiente, en condiciones en las que el flujo sanguíneo al cerebro puede verse comprometido, es imperativo evitar la actividad cerebral excesiva. Los medicamentos anticonvulsivos aumentan la inhibición neuronal o reducen los procesos excitatorios en el cerebro. La actividad epiléptica puede ir acompañada de acidosis láctica sistémica, disminución de la oxigenación arterial y aumento del dióxido de carbono; por lo tanto, es importante mantener la ventilación, la oxigenación y la presión arterial en un paciente con dicha actividad. La actividad epiléptica prolongada o recurrente puede provocar un daño cerebral profundo.
Tratamiento de la epilepsia
En pacientes con estado epiléptico, es necesario un tratamiento inmediato para detener la crisis. Se utilizan benzodiacepinas como midazolam y lorazepam para detenerla rápidamente. La administración de anticonvulsivos de mantenimiento intravenosos, como fenitoína, debe comenzar inmediatamente después de las benzodiacepinas. Si estos fármacos no logran interrumpir la actividad epileptiforme, están indicados los barbitúricos (p. ej., fenobarbital). El cambio, la administración inmediata de anticonvulsivos de mantenimiento intravenosos, fue sugerido por un neurólogo especialista en epilepsia. Representa la práctica actual. Si la crisis es de inicio reciente, es fundamental identificar y tratar las causas precipitantes corregibles, como la hipertermia, el desequilibrio electrolítico, la infección o el tumor. En combinación con la isquemia, las crisis pueden causar daño neuronal rápido y devastador, por lo que deben tratarse de forma agresiva.
Lesión Cerebral Traumática
El traumatismo es la principal causa de muerte en personas de 1 a 44 años; más de la mitad de estas muertes se deben a traumatismos craneoencefálicos. Aproximadamente 1,7 millones de lesiones cerebrales traumáticas (LCT) ocurren en Estados Unidos cada año, las cuales provocan 52 000 muertes y 1,4 millones de visitas a urgencias.
El traumatismo craneoencefálico (TCE) se clasifica comúnmente mediante la Escala de Coma de Glasgow (ECG); las puntuaciones de 13 a 15 se consideran lesión leve, de 9 a 12 lesión moderada y de 8 o menos lesión cerebral traumática grave. La fisiopatología del traumatismo craneoencefálico se divide en dos categorías: lesión cerebral primaria y lesión cerebral secundaria. Los enfoques actuales para el tratamiento del TCE se centran en las dos fases de la lesión cerebral. La lesión cerebral primaria es la interrupción directa del parénquima cerebral y su irrigación sanguínea por el traumatismo y ocurre en el momento del traumatismo. Este daño primario puede ser causado por una lesión neuronal directa por contusiones y lesión axonal difusa (LAD), herniación cerebral o la sección de vasos sanguíneos en el cerebro que produce hematoma o isquemia directa. La reversión del daño primario no es posible y, por lo tanto, no es susceptible de intervención médica. Sin embargo, gran parte del daño cerebral en pacientes traumatizados es secundario a este daño cerebral primario, y se produce como una cascada de eventos bioquímicos, celulares y moleculares que se inician en el momento del trauma inicial y continúan durante horas o días.152 Los mecanismos de daño secundario incluyen excitotoxicidad, respuestas inflamatorias, isquemia secundaria por vasoespasmo, oclusión microvascular focal y daño vascular, así como fallo energético y la consiguiente apoptosis. Estos eventos provocan la muerte neuronal, edema cerebral secundario y aumento de la presión intracraneal, lo que puede agravar aún más el daño cerebral. Este daño comparte muchas características de la cascada isquémica durante y después de un accidente cerebrovascular agudo. La entrada de calcio resultante del trauma se ha implicado como desencadenante del daño. El daño secundario puede reducirse con una monitorización y un tratamiento adecuados. La identificación, prevención y tratamiento del daño cerebral secundario son el objetivo principal del manejo en cuidados neurointensivos para pacientes con traumatismo craneoencefálico grave. El tratamiento puede incluir la reducción de la PIC (por debajo de 20 es un objetivo razonable), el mantenimiento de la presión de perfusión cerebral (PPC) (la diferencia entre la presión arterial media y la PIC), el tratamiento agresivo de la hipertermia, la evacuación de hematomas, la descompresión quirúrgica y, posiblemente, el uso de agentes farmacológicos que interfieran con la cascada de eventos que conducen al daño neuronal. La isquemia cerebral fue un hallazgo común en los estudios histológicos de casos de traumatismo terminal. Es de gran importancia prevenir la isquemia secundaria que frecuentemente sigue al traumatismo craneoencefálico y que posiblemente se deba a la liberación de sustancias vasoconstrictoras durante la reperfusión. El edema cerebral citotóxico y vasogénico suele seguir al traumatismo craneoencefálico y puede conducir a un marcado aumento de la PIC. Esto puede resultar en hipoperfusión del cerebro, incluso si se mantiene la presión arterial. La hemorragia intracraneal puede aumentar el volumen sanguíneo intracraneal y la PIC, reduciendo así la presión de perfusión cerebral. La sangre intracraneal puede causar daño al promover directamente la formación de radicales libres catalizada por el hierro de la hemoglobina. La hipotensión se ha asociado con un pronóstico mucho peor; por lo tanto, una intervención importante para mejorar el pronóstico en pacientes traumatizados es mantener una presión arterial cercana a la normal para prevenir la isquemia cerebral secundaria. Mantener una presión de perfusión cerebral (PPC) adecuada, en lugar de solo controlar la presión intracraneal (PIC), es de suma importancia; la PPC debe ser de 60 mmHg para asegurar un flujo sanguíneo cerebral óptimo. Los corticosteroides en dosis altas no son efectivos y se ha demostrado que aumentan la mortalidad por traumatismo craneoencefálico; están contraindicados según las guías de la Fundación para el Traumatismo Cerebral. Se puede recomendar el uso profiláctico a corto plazo (1 semana) de antiepilépticos (fenitoína) para la prevención de convulsiones tempranas; estos no afectan el desarrollo a largo plazo de las convulsiones y no se ha demostrado que mejoren el pronóstico.